Explain chapter hepatobillairy robbins
Hepatobiliary Chapter - Robbins & Kumar Pathologic Basis of Disease
LIVER ANATOMY (Brief Review)
- Portal vein: 60-70% of hepatic blood flow
- Hepatic artery: 30-40%
- Zone 1 (periportal) - highest O2, first affected in toxin exposure
- Zone 2 (mid-lobular)
- Zone 3 (pericentral/centrilobular) - lowest O2, first affected in ischemia and alcohol
GENERAL FEATURES OF LIVER DISEASE
Hepatocyte and Parenchymal Responses
- Steatosis (fatty change): Accumulation of fat droplets (macro- or microvesicular) due to impaired lipoprotein secretion, increased fatty acid delivery, or toxic injury
- Hepatocyte swelling (ballooning degeneration): Pale, swollen hepatocytes from disrupted cytoskeleton - hallmark of toxic or steatohepatitis
- Apoptosis: Acidophil/Councilman bodies - shrunken, eosinophilic cells
- Necrosis: Coagulative necrosis from ischemia; lytic necrosis from immune attack
- Regeneration: Hepatocytes are capable of vigorous regeneration
- Cholestasis: Intracellular and canalicular accumulation of bile pigments
Scar Formation and Regression
LIVER FAILURE
Acute Liver Failure
Chronic Liver Failure and Cirrhosis
- Micronodular cirrhosis (nodules <3 mm) - alcohol, hemochromatosis
- Macronodular cirrhosis (nodules >3 mm) - viral hepatitis, Wilson disease
- Dense collagen bands highlighted by Masson trichrome stain
VIRAL HEPATITIS
Hepatitis A (HAV)
- Fecal-oral transmission; RNA virus; never causes chronic hepatitis
- Acute self-limited illness; very rarely acute liver failure
Hepatitis B (HBV)
- DNA virus; parenteral/sexual/perinatal transmission
- Serologic markers (critical for exams):
- HBsAg: first to appear; persistence = chronic infection
- Anti-HBs: appears after resolution; confers lifelong protection; basis of vaccination
- HBeAg + HBV DNA: active viral replication, high infectivity
- Anti-HBc IgM: window period marker
- Risk of chronicity: ~90% if infected at birth, ~5-10% in adults
- Chronic infection → cirrhosis → hepatocellular carcinoma (especially in perinatal infection)
- Treatment: interferon + antivirals (entecavir, tenofovir)
Hepatitis C (HCV)
- RNA virus (Flaviviridae); blood-borne transmission
- ~80% of acutely infected individuals develop chronic infection
- Major cause of cirrhosis and HCC in the West
- Curative treatment now available: direct-acting antivirals (DAAs)
Hepatitis D (HDV)
- Defective RNA virus - requires HBsAg for assembly; only co-infects or super-infects HBV patients
- Superinfection with HDV accelerates liver disease
Hepatitis E (HEV)
- Fecal-oral; RNA virus; endemic in Asia/Africa
- Particularly severe in pregnant women (mortality up to 20%)
- Generally self-limited but can cause acute liver failure
AUTOIMMUNE HEPATITIS
- Middle-aged women predominantly; associated with other autoimmune diseases (type 1 DM, thyroiditis, celiac disease)
- Type 1: ANA + anti-smooth muscle actin (anti-SMA) - most common
- Type 2: Anti-LKM-1 antibodies (anti-CYP2D6) - more common in children
- Histology: interface hepatitis + prominent plasma cell infiltrate (key feature)
- Diagnosis: simplified scoring system (autoantibodies + IgG elevation + liver biopsy + exclusion of other causes)
- Treatment: prednisone ± azathioprine → remission in 80-90%
DRUG- AND TOXIN-INDUCED LIVER INJURY (DILI)
- Predictable (dose-dependent): acetaminophen (zone 3 necrosis) - #1 cause of acute liver failure in West
- Unpredictable (idiosyncratic): isoniazid, halothane, sulfonamides
- Morphologic patterns: steatosis, cholestasis, hepatitis, vascular lesions, granulomas
- Amiodarone and tamoxifen can cause steatohepatitis resembling MASLD
STEATOTIC LIVER DISEASE (MASLD / MASH)
- Mixed macro- and microvesicular steatosis
- Ballooned hepatocytes
- Lobular inflammation (neutrophils and lymphocytes)
- "Chicken wire" or pericellular fibrosis - sinusoidal fibrosis pattern
- Mallory-Denk bodies (same as alcohol-related liver disease)
INHERITED LIVER DISEASES
Hemochromatosis
- Hereditary hemochromatosis: autosomal recessive; most common mutation = HFE gene (C282Y)
- Iron accumulates in hepatocytes, pancreas, heart, joints, skin, gonads
- Liver: micronodular cirrhosis + brown pigment (hemosiderin) in hepatocytes
- Perl's Prussian blue stain: blue iron deposits
- Clinical: "Bronze diabetes" - cirrhosis + diabetes + skin hyperpigmentation
- Increased risk of HCC (200x), cardiomyopathy, arthropathy
- Treatment: phlebotomy
Wilson Disease
- Autosomal recessive; mutation in ATP7B (copper-transporting ATPase)
- Copper accumulates in liver, brain (basal ganglia - choreoathetosis), eyes (Kayser-Fleischer rings), kidneys
- Liver: fatty change → hepatitis → cirrhosis; acute liver failure possible
- Lab: low serum ceruloplasmin, elevated urinary copper
- Treatment: D-penicillamine or trientine
Alpha-1 Antitrypsin Deficiency
- Autosomal recessive; misfolded PiZZ protein accumulates in ER of hepatocytes
- Liver: PAS-positive, diastase-resistant globules in periportal hepatocytes
- Clinical: neonatal hepatitis, cirrhosis in children/adults; emphysema in lungs
Neonatal Cholestasis
- Most commonly idiopathic (neonatal giant cell hepatitis)
- Biliary atresia: fibro-obliterative disease of extrahepatic bile ducts - emergency surgical repair (Kasai procedure)
CHOLESTASIS
- Intrahepatic: hepatocyte dysfunction, drug toxicity, viral hepatitis, pregnancy, PBC, PSC
- Extrahepatic (obstructive): gallstones, pancreatic carcinoma, cholangiocarcinoma, strictures
- Crigler-Najjar type 1: severe UGT1A1 deficiency → fatal unconjugated hyperbilirubinemia
- Crigler-Najjar type 2: partial UGT1A1 deficiency → manageable
- Gilbert syndrome: mild UGT1A1 deficiency → benign unconjugated hyperbilirubinemia (stress/fasting triggered)
- Dubin-Johnson syndrome: MRP2 mutation → conjugated hyperbilirubinemia + black liver pigment (benign)
- Rotor syndrome: conjugated hyperbilirubinemia, no pigment (benign)
Primary Biliary Cholangitis (PBC)
- Middle-aged women; anti-mitochondrial antibodies (AMA) in >90%
- Autoimmune destruction of small intrahepatic bile ducts; granulomatous cholangitis (florid duct lesion)
- Associated with Sjögren syndrome and Hashimoto thyroiditis
- Progressive → biliary cirrhosis; treatment: ursodeoxycholic acid
Primary Sclerosing Cholangitis (PSC)
- Middle-aged men; strongly associated with ulcerative colitis (70%)
- Inflammation, fibrosis, and "beaded" stricturing of large intra- and extrahepatic bile ducts on MRCP/ERCP
- Histology: "onion-skin" periductal fibrosis
- Elevated IgG4 in subset (IgG4-related PSC variant)
- Risk of cholangiocarcinoma (10-15% lifetime risk)
HEPATIC VASCULAR DISEASES
- Budd-Chiari syndrome: thrombosis of hepatic veins → centrilobular congestion and necrosis → congestive cirrhosis; associated with hypercoagulable states (polycythemia vera)
- Sinusoidal obstruction syndrome (SOS)/Veno-occlusive disease: injury to sinusoidal endothelium → fibrous obliteration of terminal hepatic venules; seen after bone marrow transplant (chemotherapy-related)
- Portal vein thrombosis: cirrhosis, hypercoagulable states → portal hypertension without cirrhosis
- Peliosis hepatis: blood-filled sinusoidal cavities; associated with anabolic steroids, OCP, Bartonella
HEPATIC NODULES AND TUMORS
Benign - Focal Nodular Hyperplasia (FNH)
- Most common in women (but NOT hormone-driven)
- Not a true neoplasm; thought to arise from aberrant blood flow
- Central stellate scar in up to 80% of cases with radiating fibrous septa
- No risk of malignant transformation; no rupture
Benign - Hepatocellular Adenoma
- True benign neoplasm; strongly associated with oral contraceptive use (estrogen)
- Four subtypes based on driver mutations (HNF1A, beta-catenin, IL-6 pathway)
- Risk of bleeding (rupture) especially if >5 cm
- Beta-catenin mutated subtype has risk of malignant transformation to HCC
- Treatment: stop OCP + surgical resection if large/symptomatic
Malignant - Hepatocellular Carcinoma (HCC)
- Most common primary liver malignancy worldwide
- Risk factors: chronic HBV (most common globally), chronic HCV, alcoholic cirrhosis, MASLD/MASH, hemochromatosis, aflatoxin B1 (from Aspergillus in contaminated grain)
- Pathogenesis: chromosomal instability → mutations in TP53, CTNNB1 (beta-catenin), TERT promoter
- Morphology: trabecular pattern (most common), pseudoglandular, or solid pattern; bile production by tumor cells is pathognomonic
- AFP (alpha-fetoprotein): elevated in ~75%; useful for surveillance in high-risk patients
- Fibrolamellar HCC: variant in young patients without cirrhosis; DNAJB1-PRKACA fusion; better prognosis
Malignant - Intrahepatic Cholangiocarcinoma
- Arises from intrahepatic bile duct epithelium (cholangiocytes)
- Risk factors: PSC, liver flukes (Clonorchis sinensis, Opisthorchis), Caroli disease, hepatolithiasis
- Histology: adenocarcinoma with desmoplastic stroma; perineural invasion
- Poor prognosis; often unresectable at diagnosis
Metastatic Liver Disease
- Far more common than primary tumors
- Common primary sources: colon (#1), breast, lung, pancreas
- Multiple nodular deposits; massive hepatomegaly; can replace most of parenchyma with minimal liver dysfunction until late
GALLBLADDER
Cholelithiasis (Gallstones)
- Cholesterol stones (75% in Western countries): pale yellow; form when bile becomes supersaturated with cholesterol relative to bile salts and lecithin; risk factors = "5 Fs" - Fat, Forty, Female, Fertile, Fair; also ileal disease (Crohn), rapid weight loss, drugs (OCPs, clofibrate)
- Pigment stones: black stones (hemolytic anemias - excess unconjugated bilirubin) vs. brown stones (biliary infections/parasites)
Acute Cholecystitis
- 90% from gallstone obstruction of cystic duct → bile stasis → prostaglandin-mediated inflammation
- Acalculous cholecystitis: in critically ill patients (ICU), burns, trauma, post-op
- Morphology: distended, hyperemic, edematous wall; neutrophilic infiltrate; possible perforation and gangrenous changes
Chronic Cholecystitis
- Very common; nearly always associated with gallstones
- Rokitansky-Aschoff sinuses: outpouchings of gallbladder mucosa into the wall (from increased intraluminal pressure)
- Porcelain gallbladder: dystrophic calcification of the wall (historically linked to gallbladder carcinoma risk)
Gallbladder Carcinoma
- Uncommon; strongly associated with gallstones (~90%)
- More common in women; usually adenocarcinoma
- Detected late (often incidentally after cholecystectomy for stones or at autopsy)
- Very poor prognosis: 5-year survival <5% for advanced disease
- Spreads directly to liver; lymph nodes at porta hepatis
Structural Anomalies of the Biliary Tree
- Choledochal cysts: congenital dilatation of common bile duct; risk of cholangiocarcinoma; female predominance
- Fibropolycystic disease: ductal plate malformation spectrum; includes von Meyenburg complexes (bile duct hamartomas), Caroli disease (saccular intrahepatic duct dilatation), congenital hepatic fibrosis
HEPATIC DISEASE IN PREGNANCY
- Preeclampsia/Eclampsia: periportal fibrin deposits, ischemic hepatocyte necrosis; HELLP syndrome (Hemolysis, Elevated Liver enzymes, Low Platelets)
- Acute fatty liver of pregnancy (AFLP): third trimester; microvesicular steatosis of hepatocytes; mitochondrial defect (LCHAD mutation); can progress rapidly to liver failure; treatment = deliver the baby
- Intrahepatic cholestasis of pregnancy (ICP): pruritus in 2nd/3rd trimester; elevated bile salts; canalicular cholestasis; resolves after delivery; risk of fetal loss
SUMMARY TABLE: KEY HIGH-YIELD ASSOCIATIONS
| Disease | Key Feature / Marker |
|---|---|
| Hereditary hemochromatosis | HFE C282Y mutation; Prussian blue stain; "bronze diabetes" |
| Wilson disease | ATP7B mutation; Kayser-Fleischer rings; low ceruloplasmin |
| Alpha-1-antitrypsin def. | PAS+, diastase-resistant globules in hepatocytes |
| PBC | AMA; granulomatous duct destruction; middle-aged women |
| PSC | UC association; "onion skin" fibrosis; beaded ducts on MRCP |
| HCC | AFP elevated; trabecular pattern; aflatoxin B1 |
| Fibrolamellar HCC | Young adults, no cirrhosis; DNAJB1-PRKACA fusion |
| Cholangiocarcinoma | Liver flukes; PSC; desmoplastic; perineural invasion |
| FNH | Central stellate scar; no malignant potential |
| Hepatocellular adenoma | OCP-related; risk of rupture and HCC (beta-cat subtype) |
| DILI (acetaminophen) | Zone 3 necrosis; #1 acute liver failure in West |
| Acute fatty liver of pregnancy | Microvesicular steatosis; deliver immediately |
| Dubin-Johnson syndrome | MRP2 mutation; black liver; conjugated hyperbilirubinemia |
| Budd-Chiari | Hepatic vein thrombosis; centrilobular necrosis |
Make a comparison poster of viral hepatitis (A-E) serology and outcomes
A detailed medical comparison poster titled "Viral Hepatitis A–E: Serology & Outcomes" for medical students. The poster has a dark navy blue background with white and colored text. It is organized as a clean comparison table with 6 columns (one per virus: HAV, HBV, HCV, HDV, HEV) and rows covering: Virus Type, Transmission, Incubation Period, Key Serologic Markers, Chronicity, Fulminant Failure Risk, Vaccine Available, and Clinical Outcome/Notes. Content for each virus: HAV column (yellow accent): - Virus: RNA, Picornavirus - Transmission: Fecal-oral - Incubation: 15–45 days - Serology: Anti-HAV IgM (acute), Anti-HAV IgG (immunity/past) - Chronicity: NEVER - Fulminant: Rare (<0.1%) - Vaccine: YES - Outcome: Self-limited; no chronic disease HBV column (orange accent): - Virus: DNA, Hepadnavirus - Transmission: Parenteral, sexual, perinatal - Incubation: 30–180 days - Serology: HBsAg (infection), Anti-HBs (immunity), HBeAg (active replication), Anti-HBc IgM (acute), HBV DNA - Chronicity: 5–10% adults; 90% neonates - Fulminant: 0.1–0.5% - Vaccine: YES - Outcome: Chronic → cirrhosis → HCC risk HCV column (red accent): - Virus: RNA, Flavivirus - Transmission: Parenteral (blood) - Incubation: 15–160 days - Serology: Anti-HCV (exposure), HCV RNA (active infection), Genotyping (treatment) - Chronicity: ~80% - Fulminant: Very rare - Vaccine: NO - Outcome: High chronic rate → cirrhosis → HCC; curable with DAAs HDV column (purple accent): - Virus: Defective RNA (requires HBsAg) - Transmission: Parenteral, sexual (only with HBV) - Incubation: Same as HBV - Serology: Anti-HDV IgM/IgG, HDV RNA; Co-infection vs Superinfection - Chronicity: Co-inf: 2–5%; Superinfection: 70–80% - Fulminant: High (superinfection) - Vaccine: HBV vaccine prevents HDV - Outcome: Accelerates HBV disease; worst prognosis HEV column (green accent): - Virus: RNA, Hepevirus - Transmission: Fecal-oral (contaminated water) - Incubation: 15–60 days - Serology: Anti-HEV IgM (acute), Anti-HEV IgG (past/immunity), HEV RNA - Chronicity: Rare (immunocompromised) - Fulminant: HIGH in pregnancy (10–20%) - Vaccine: YES (China only) - Outcome: Self-limited; DANGEROUS in pregnancy Below the table, include a color-coded legend bar and a small footer note: "Source: Robbins & Kumar Pathologic Basis of Disease". The poster should be professional, readable, and visually structured like a high-quality medical education infographic with clear column dividers and row alternating shading.
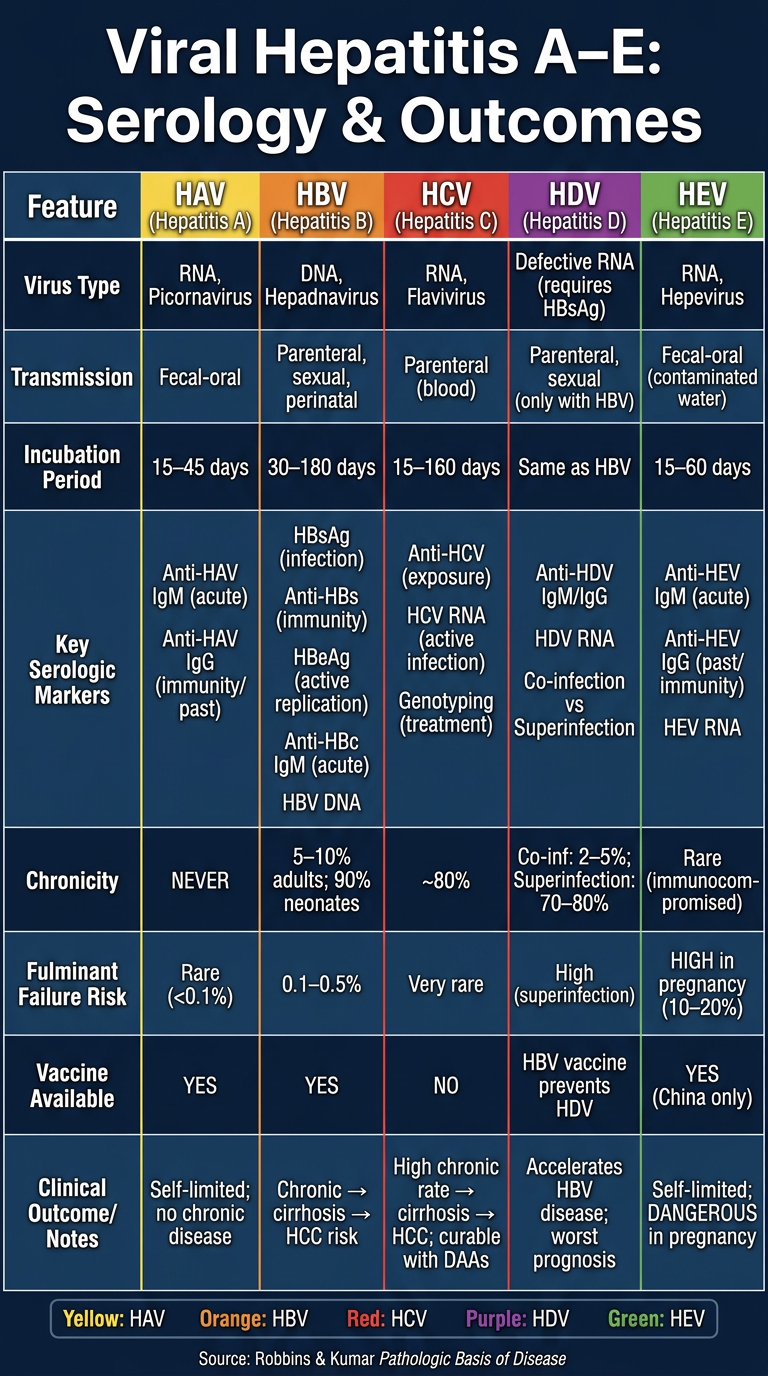
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Virus type | RNA | DNA | RNA | Defective RNA | RNA |
| Transmission | Fecal-oral | Parenteral/sexual/perinatal | Parenteral | With HBV only | Fecal-oral |
| Key markers | Anti-HAV IgM/IgG | HBsAg, HBeAg, Anti-HBs, Anti-HBc IgM | Anti-HCV, HCV RNA | Anti-HDV, HDV RNA | Anti-HEV IgM/IgG |
| Chronicity | Never | 5-10% adults; 90% neonates | ~80% | Up to 80% (superinfection) | Rare |
| Fulminant risk | Rare | 0.1-0.5% | Very rare | High (superinfection) | 10-20% in pregnancy |
| Vaccine | Yes | Yes | No | HBV vaccine prevents it | Yes (China only) |
| Outcome | Self-limited | Cirrhosis, HCC risk | Cirrhosis, HCC; curable with DAAs | Worst HBV accelerator | Safe except in pregnancy |
- HCV has the highest chronicity rate (~80%) and no vaccine
- HDV needs HBsAg to survive - vaccinating against HBV prevents HDV
- HEV is uniquely dangerous in pregnant women (20% mortality)
- HBV is the only DNA virus among the five
- Only HAV and HEV are fecal-oral; both are typically self-limited