Thrombotic thrombocytopenia
Reading File
Finding Sources
Searching PubMed
"thrombotic thrombocytopenic purpura" AND treatment
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Searching Images
thrombotic thrombocytopenic purpura schistocytes blood smear TTP
Now I have comprehensive information to provide a thorough answer on TTP. Let me compile the response.
Thrombotic Thrombocytopenic Purpura (TTP)
Overview
TTP is the most important primary thrombotic microangiopathy (TMA) — a group of disorders characterized by microangiopathic hemolytic anemia (MAHA) and thrombocytopenia, caused by microvascular thrombosis in arterioles and capillaries that simultaneously destroys RBCs and consumes platelets.
Pathogenesis
The central defect is deficiency of, or autoantibodies against, ADAMTS13 — a plasma metalloproteinase (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) that normally cleaves ultra-large von Willebrand factor (VWF) multimers secreted by endothelial cells into smaller, less adhesive fragments.
When ADAMTS13 is absent or inhibited:
- Ultra-large VWF multimers persist in circulation
- These bind platelets avidly via glycoprotein Ib, forming massive VWF-platelet aggregates
- Microthrombi deposit in small vessels → tissue ischemia, platelet consumption, and mechanical RBC destruction (schistocytes)
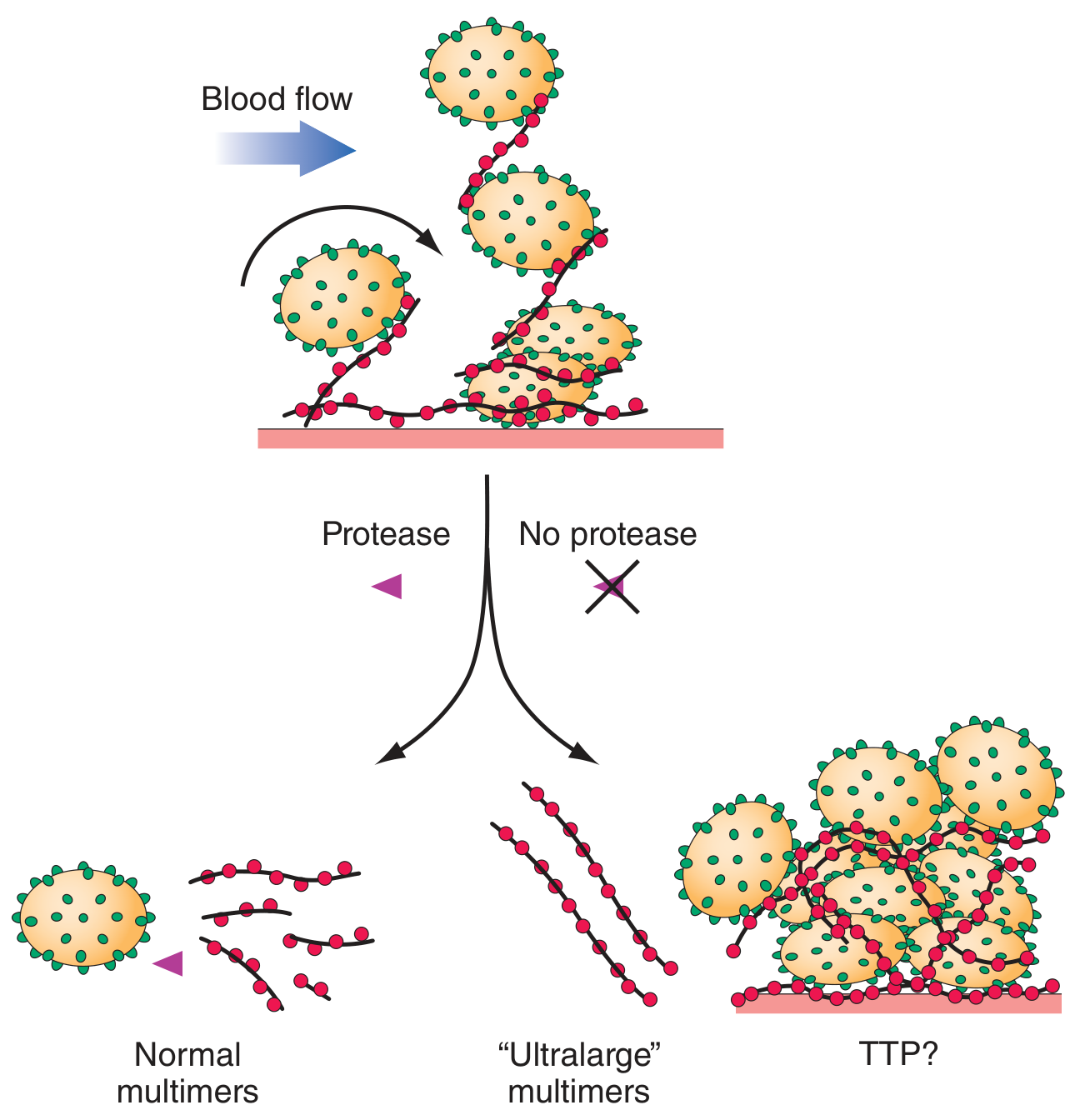
This defect alone is not sufficient — a "two-hit" model applies: ADAMTS13 deficiency predisposes, but a triggering event (infection, pregnancy) activates endothelial cells and precipitates the episode. — Comprehensive Clinical Nephrology, 7th Ed.
ADAMTS13 activity <10% is diagnostic of TTP. — Harrison's 22e
Forms
| Form | Mechanism |
|---|---|
| Acquired (immune-mediated, ~95%) | IgG autoantibodies to ADAMTS13; associated with HIV, pregnancy, SLE, drugs (ticlopidine, clopidogrel) |
| Hereditary (Upshaw-Schulman syndrome) | Congenital ADAMTS13 gene mutations; episodic, often triggered by first pregnancy |
| Drug-induced (non-immune) | Direct endothelial toxicity: cyclosporine, gemcitabine, mitomycin C, tacrolimus |
Classic Pentad (not always complete)
Historically described as five features — though the full pentad is rarely seen together:
- Thrombocytopenia (platelet count often <30 × 10⁹/L)
- Microangiopathic hemolytic anemia (MAHA) — schistocytes on peripheral smear
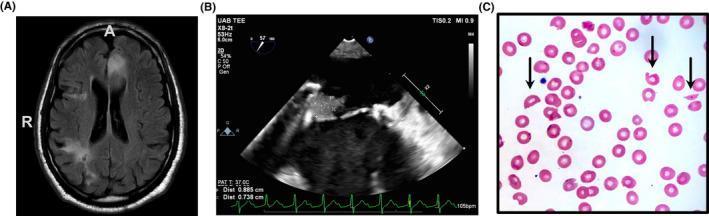
- Neurologic symptoms — fluctuating mental clouding, confusion, stroke (most distinguishing from HUS)
- Renal dysfunction — mainly proteinuria/hematuria; severe AKI is uncommon (unlike HUS)
- Fever
Cardiac (25%) and mesenteric ischemia (35%, presenting as abdominal pain/diarrhea) are also recognized. — Rosen's Emergency Medicine
Clinical Image: Peripheral Blood Smear & Multi-organ Involvement
Epidemiology
- First acute episode: ~90% during adulthood
- ~2× more common in women
- Increased risk in: HIV infection, pregnancy, autoimmune disease
Diagnosis
Key Labs
| Test | Finding in TTP |
|---|---|
| CBC | Thrombocytopenia |
| Peripheral smear | Schistocytes (helmet cells) |
| LDH | Elevated (hemolysis + tissue ischemia) |
| Haptoglobin | Undetectable |
| Indirect bilirubin | Elevated |
| PT/aPTT | Usually normal (distinguishes from DIC) |
| Creatinine | Mildly elevated or normal |
| ADAMTS13 activity | <10% = diagnostic |
| Direct antiglobulin (DAT) | Negative (rules out autoimmune hemolytic anemia) |
PLASMIC Score (for identifying candidates for urgent plasma exchange)
| Criterion | Points |
|---|---|
| Platelet count <30 × 10⁹/L | +1 |
| Hemolysis (retic >2.5%, haptoglobin undetectable, or indirect bili >2.0 mg/dL) | +1 |
| Active cancer or treated within past year | 0 (vs. +1 if no cancer) |
| Solid organ or stem cell transplant history | 0 (vs. +1 if none) |
| MCV <90 fL | +1 |
| INR <1.5 | +1 |
| Creatinine <2.0 mg/dL | +1 |
Score 0–4: Low risk — consider alternative diagnosis
Score 5: Intermediate — consult hematology, consider plasma exchange
Score 6–7: High risk — immediate plasma exchange — Rosen's Emergency Medicine
Treatment
1. Therapeutic Plasma Exchange (TPE) — Mainstay
- Replaces deficient ADAMTS13 and removes anti-ADAMTS13 autoantibodies
- Continued daily until platelet count normalizes (≥150,000/μL) and hemolysis resolves for ≥2 days
- Reduced mortality from 85–100% → 10–30% — Harrison's 22e
- Fresh-frozen plasma (FFP) and cryosupernatant are equivalent
2. Glucocorticoids
- Adjunct to TPE; suppress autoantibody production
- Prednisone 1 mg/kg/day PO, or methylprednisolone 1 g IV × 3 days for severe/neurologic cases
- Never sufficient alone
3. Caplacizumab (anti-VWF nanobody) — Key addition
- Blocks interaction between VWF multimers and platelet GP Ib
- Dose: 11 mg IV loading, then 11 mg SC daily for ≥10 days
- Phase 3 HERCULES trial: shorter time to platelet normalization, reduced thromboembolic events, death, and relapse
- ISTH guidelines: recommended in patients with ADAMTS13 <10% or high clinical probability
- Important: does not affect ADAMTS13 autoantibody production — TPE + immunosuppression still required — Comprehensive Clinical Nephrology, 7th Ed.
Recent meta-analysis (PMID: 38874905, 2024): Systematic review confirms efficacy and relative safety of caplacizumab in immune-mediated TTP.
4. Rituximab (anti-CD20)
- Indicated for: refractory disease, relapse prevention, or patients with persistent anti-ADAMTS13 inhibitors
- Induces clinical remission, clears anti-ADAMTS13 antibodies, raises ADAMTS13 activity >10%
- Remission duration: 9 months–4 years; ~10% relapse rate
- ISTH guidelines: use when inhibitor is confirmed
5. Platelet Transfusion — AVOID
- Contraindicated (except life-threatening bleeding) — additional platelets fuel microvascular thrombosis
6. Hereditary TTP (Upshaw-Schulman)
- Plasma infusion (not exchange) replaces deficient ADAMTS13
- Prophylactic plasma infusions every 2–3 weeks for frequent relapsers
- Recombinant ADAMTS13 — showing efficacy in ongoing trials (PMID: 41934124, 2026 meta-analysis)
TTP vs. HUS
| Feature | TTP | HUS |
|---|---|---|
| Predominant age | Adults | Children (often post-GI illness) |
| Neurologic involvement | Prominent | Mild |
| Renal involvement | Mild (proteinuria/hematuria) | Severe AKI (prominent) |
| ADAMTS13 | <10% | Usually normal |
| Etiology | ADAMTS13 deficiency/inhibitor | Shiga toxin (STEC), complement dysregulation |
VITT (Vaccine-Induced Immune Thrombotic Thrombocytopenia)
A distinct syndrome (post-adenoviral vector COVID-19 vaccines) with features overlapping TTP but caused by anti-PF4 antibodies (similar mechanism to HIT). Characterized by high D-dimer, thrombosis at unusual sites (cerebral venous sinuses), ~20% fatal. Treatment: IVIgG + non-heparin anticoagulant. Heparin is contraindicated. — Harrison's 22e
Sources: Harrison's Principles of Internal Medicine 22e; Rosen's Emergency Medicine; Comprehensive Clinical Nephrology 7th Ed.; Brenner & Rector's The Kidney
TTP course
Reading File
Finding Sources
Finding Sources
Searching PubMed
"thrombotic thrombocytopenic purpura" AND relapse
Finding Sources
Reading Article
Reading File
Reading File
Reading File
Finding Sources
I now have comprehensive information on the clinical course of TTP. Here's a full synthesis:
TTP: Clinical Course
Acute Episode
Onset & Early Symptoms
The initial presentation may be deceptively mild. Early symptoms include:
- Fatigue, nausea, vomiting, dyspnea
- Petechiae, bruising
As microvascular thrombosis progresses, end-organ ischemia develops:
| Organ | Manifestations |
|---|---|
| CNS (most characteristic) | Headache, confusion, weakness → seizures, stroke, coma |
| GI | Abdominal pain, diarrhea — present in ~50% |
| Cardiac | Myocardial infarction; arrhythmias are a frequent cause of death |
| Renal | Mild: proteinuria/hematuria; creatinine typically 1.7–2.3 mg/dL — severe AKI is uncommon |
| Hematologic | Thrombocytopenia (often <30 × 10⁹/L), MAHA with schistocytes |
Without Treatment
Historically, TTP was almost universally fatal — mortality 85–100% within weeks to months. — Harrison's 22e; Brenner & Rector's The Kidney
Response to Treatment
With therapeutic plasma exchange (TPE):
- Mortality falls to ~10–30% — Harrison's 22e
- Remission (sustained platelet normalization + resolution of hemolysis) achieved in >80% — Tintinalli's EM
- With modern triple therapy (TPE + corticosteroids + rituximab): 30-day survival >90% — JAMA 2025, PMID 40388146
- TPE is continued daily until platelets ≥150,000/μL for ≥2 consecutive days and hemolysis resolves
Refractory TTP (failure to respond to daily TPE): TPE may be escalated to twice daily; rituximab or cyclosporine added.
Remission
Clinical remission is defined as:
- ≥30 days of sustained platelet normalization
- Decreased LDH
- Absence of new or progressive ischemic organ injury
- Without TPE or caplacizumab
ADAMTS13 activity typically recovers above 10%, though it may remain low — a persistent level <20% in remission is a red flag for impending relapse.
Relapse
Relapse is defined as a new TTP episode >30 days after completing remission-achieving TPE.
| Parameter | Data |
|---|---|
| Overall relapse rate (historical) | 20–50% — Tintinalli's EM |
| Rate with rituximab use | Falling — previously ~33%; rituximab reduces risk significantly |
| Timing | ~50% of relapses within the first year; most within 2 years — Brenner & Rector's |
| Risk factor | ADAMTS13 activity <10% = highest relapse risk |
| Rituximab prophylaxis in remission | When ADAMTS13 <20% detected: rituximab reduces relapse risk (OR 0.09, 95% CI 0.04–0.24) — JAMA 2025 |
Features of Relapses
- May present with milder symptoms and less severe hematologic findings than the initial episode
- Patients with multiple relapses often experience them at progressively longer intervals
- Subsequent episodes typically require less total plasma volume and shorter TPE duration — Tintinalli's EM
- Triggers are often the same as the original episode (infection, pregnancy, drugs)
Special Situations Affecting Course
Pregnancy
- ADAMTS13 activity decreases by up to 30% physiologically during pregnancy
- TTP onset is most common in the second or third trimester (before 24 weeks should raise suspicion vs. preeclampsia/HELLP)
- Maternal mortality has decreased significantly with TPE; however, fetal mortality remains high due to placental microvascular occlusion and infarction
- Women with prior iTTP have risk of relapse with subsequent pregnancies — serial ADAMTS13 monitoring every trimester recommended — Tintinalli's EM; Creasy & Resnik's MFM
HIV-associated TTP
- HIV progression to AIDS is a recognized precipitant of MAHA and thrombocytopenia
- Course follows general TTP pattern; treating the underlying HIV may reduce recurrences
Drug-associated TTP
- Immune-mediated (ticlopidine, possibly clopidogrel): autoantibody-driven; days to weeks to achieve remission after drug withdrawal; ticlopidine causes more severe thrombocytopenia/hemolysis
- Endothelial toxic (cyclosporine, gemcitabine, mitomycin C, tacrolimus): dose reduction or withdrawal is primary treatment; generally not responsive to TPE
Hereditary TTP (Upshaw-Schulman)
- Episodic course; triggered by pregnancy, infection, inflammation
- Treated with plasma infusion rather than exchange during acute episodes
- Patients with frequent relapses require prophylactic plasma infusions every 2–3 weeks; maintain ADAMTS13 at ≥5% activity — Comprehensive Clinical Nephrology 7th Ed.
Monitoring in Remission
- Regular ADAMTS13 activity measurement
- If ADAMTS13 falls <20% → pre-emptive rituximab to prevent relapse (JAMA 2025, PMID 40388146)
- Ongoing vigilance for relapse triggers (infections, new medications, pregnancy planning)
Summary of Key Prognostic Figures
| Milestone | Outcome |
|---|---|
| Untreated mortality | 85–100% |
| With TPE alone | ~10–30% mortality |
| With TPE + steroids + rituximab (modern) | >90% 30-day survival |
| Relapse rate (overall) | 20–50% |
| Relapse within 1st year | ~50% of all relapses |
| Relapse with ADAMTS13 monitoring + rituximab prophylaxis | Substantially reduced (OR ~0.09) |
Sources: Tintinalli's Emergency Medicine; Brenner & Rector's The Kidney; Comprehensive Clinical Nephrology 7th Ed.; Harrison's Principles of Internal Medicine 22e; JAMA 2025 (PMID 40388146 — Pishko, Li, Cuker)
TTP latest updates
Reading File
Searching PubMed
"thrombotic thrombocytopenic purpura" AND treatment
Searching the Web
TTP thrombotic thrombocytopenic purpura 2025 2026 new treatment guidelines updates
Reading Article
Reading Article
Reading Article
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.