1. Discuss myelodysplastic syndrome. Discuss their pathophysiology, laboratory investigations, and prognostic markers. Add note to current concepts in MDS. 2. Discuss definition, classification, pathophysiology, and laboratory findings of myeloproliferative disorders. Discuss its differential diagnosis with supportive laboratory investigations.
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Searching PubMed
myelodysplastic syndrome molecular pathogenesis treatment 2024
Reading File
Reading File
Searching PubMed
myelodysplastic syndrome current treatment imetelstat luspatercept
Reading File
Reading File
Loading Image
Reading File
PART 1: MYELODYSPLASTIC SYNDROME (MDS)
Definition and Overview
Myelodysplastic syndrome (MDS) refers to a heterogeneous group of clonal stem cell disorders characterized by ineffective hematopoiesis, morphologic dysplasia of one or more myeloid cell lines, peripheral blood cytopenias, and a variable risk of transformation to acute myeloid leukemia (AML). It occurs primarily in persons over age 50 (median age at diagnosis: seventh to eighth decade). In the United States, it affects up to 15,000 people per year - roughly as common as AML. Historically termed "preleukemia" or "dysmyelopoietic syndrome," MDS is now recognized as a clonal neoplastic process. - Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 740; Robbins Basic Pathology, p. 405
Pathophysiology
1. Clonal Origin and Stem Cell Defect
MDS arises from a single mutant multipotent hematopoietic stem cell (HSC). The disorder represents a failure of normal differentiation coupled with accelerated apoptosis of maturing cells - the result is a cellular marrow producing very few functional blood elements (ineffective hematopoiesis). - Henry's, p. 740
2. Genetics and Molecular Mutations
Molecular analyses show that MDS develops through accumulation of oncogenic "driver" mutations. Most patients carry 2-3 driver mutations in:
- RNA splicing genes: SF3B1 (strongly associated with ring sideroblasts), SRSF2, U2AF1, ZRSR2
- DNA methylation genes: TET2, DNMT3A, IDH1/2
- Chromatin modification genes: ASXL1, EZH2
- Transcription regulators: RUNX1
- DNA repair: TP53 (associated with complex karyotype; worst prognosis)
- Signal transduction: CBL, NRAS, KRAS
- Cohesin complex: STAG2
Abnormalities of the spliceosome and DNA methylation lead to epigenetic disruption, altering gene expression without direct DNA mutation. - Henry's, p. 740
3. Cytogenetic Abnormalities
Cytogenetic changes are found in 40-70% of de novo MDS. Key abnormalities:
| Cytogenetic Finding | Risk Category |
|---|---|
| del(5q), isolated del(20q), -Y, normal karyotype | Low risk (favorable) |
| Trisomy 8, del(7q), isolated abnormalities | Intermediate risk |
| Monosomy 7, monosomy 5, complex karyotype (≥3 abnormalities) | High risk (unfavorable) |
Trisomy 8, monosomy 7, del(7q), monosomy 5, del(5q), del(20q), and complex karyotypes are the most commonly encountered. - Henry's, p. 737
4. Mechanisms of Cytopenia
Despite a hypercellular marrow, cytopenias arise from:
- Accelerated intramedullary apoptosis of abnormal hematopoietic precursors
- Dysmaturation with functional defects in surviving cells
- Immune-mediated suppression (especially in hypoplastic MDS)
- Overactivation of TGF-β signaling inhibiting late-stage erythropoiesis (the basis for luspatercept therapy)
5. Secondary (Therapy-Related) MDS
Secondary MDS follows prior chemotherapy (especially alkylating agents and topoisomerase II inhibitors) or radiation. It carries a worse prognosis, often with complex cytogenetics and TP53 mutations.
WHO Classification (2016/2022)
| Category | Key Features |
|---|---|
| MDS with single lineage dysplasia (MDS-SLD) | Dysplasia in ≥10% of one lineage; cytopenias; <5% BM blasts; <1% blood blasts |
| MDS with ring sideroblasts (MDS-RS-SLD/MLD) | ≥15% ring sideroblasts (or ≥5% if SF3B1 mutation present); erythroid dysplasia |
| MDS with multilineage dysplasia (MDS-MLD) | Dysplasia in ≥10% of two or more lineages; <5% BM blasts |
| MDS with excess blasts-1 (MDS-EB-1) | 5-9% BM blasts or 2-4% blood blasts |
| MDS with excess blasts-2 (MDS-EB-2) | 10-19% BM blasts or 5-19% blood blasts; Auer rods may be present |
| MDS with isolated del(5q) | Isolated del(5q); anemia; platelets normal or elevated; favorable prognosis |
| MDS, unclassifiable (MDS-U) | Pancytopenia with single lineage dysplasia; specific cytogenetic abnormality present |
Note: The 5th Edition WHO 2022 classification has consolidated and renamed several categories, moving toward molecular-based classification. MDS-EB-2 now borders AML (≥20% blasts = AML). The concept of "MDS with mutated SF3B1" is now an independent category.
- Henry's Clinical Diagnosis, pp. 3329-3356
Laboratory Investigations
A. Peripheral Blood
- CBC: Normocytic or macrocytic anemia (most common), often with reticulocytopenia. Neutropenia and/or thrombocytopenia in varying degrees (cytopenias of one to three lineages)
- Peripheral smear:
- Oval macrocytes, anisocytosis, poikilocytosis
- Pseudo-Pelger-Huët cells (bilobed/monolobed neutrophils with hypogranulation) - pathognomonic of dysgranulopoiesis
- Giant or hypogranular platelets; micro-megakaryocytes
- Blasts rarely present in early MDS (<1% in low-risk)
- Basophilic stippling; dacryocytes
B. Bone Marrow Examination
The cornerstone of diagnosis:
- Aspirate: Usually normocellular to hypercellular (rarely hypocellular ~10%)
- Dysplastic changes in ≥10% of cells in one or more lineages:
Dyserythropoiesis:
- Megaloblastoid changes, nuclear fragmentation/karyorrhexis
- Multinuclearity, nuclear bridging, binucleation
- Ring sideroblasts (iron-laden mitochondria encircling ≥1/3 of nucleus; seen on Prussian blue stain)
- Basophilic stippling
Dysgranulopoiesis:
- Nuclear-cytoplasmic asynchrony
- Hypogranulation, nuclear hyposegmentation (pseudo-Pelger-Huët)
- Occasional large azurophilic granules
- Monocyte-like appearance of neutrophil precursors
Dysmegakaryocytopoiesis:
-
Micromegakaryocytes (smallest dysplastic element)
-
Large megakaryocytes with unsegmented (monolobed) or multiple small separated nuclei
-
Decreased megakaryocyte count possible
-
Trephine biopsy: Assesses overall cellularity, fibrosis (reticulin stain), blast clusters (ALIP - Abnormal Localization of Immature Precursors)
-
Blast count: Critical for classification (<5%, 5-9%, 10-19%)
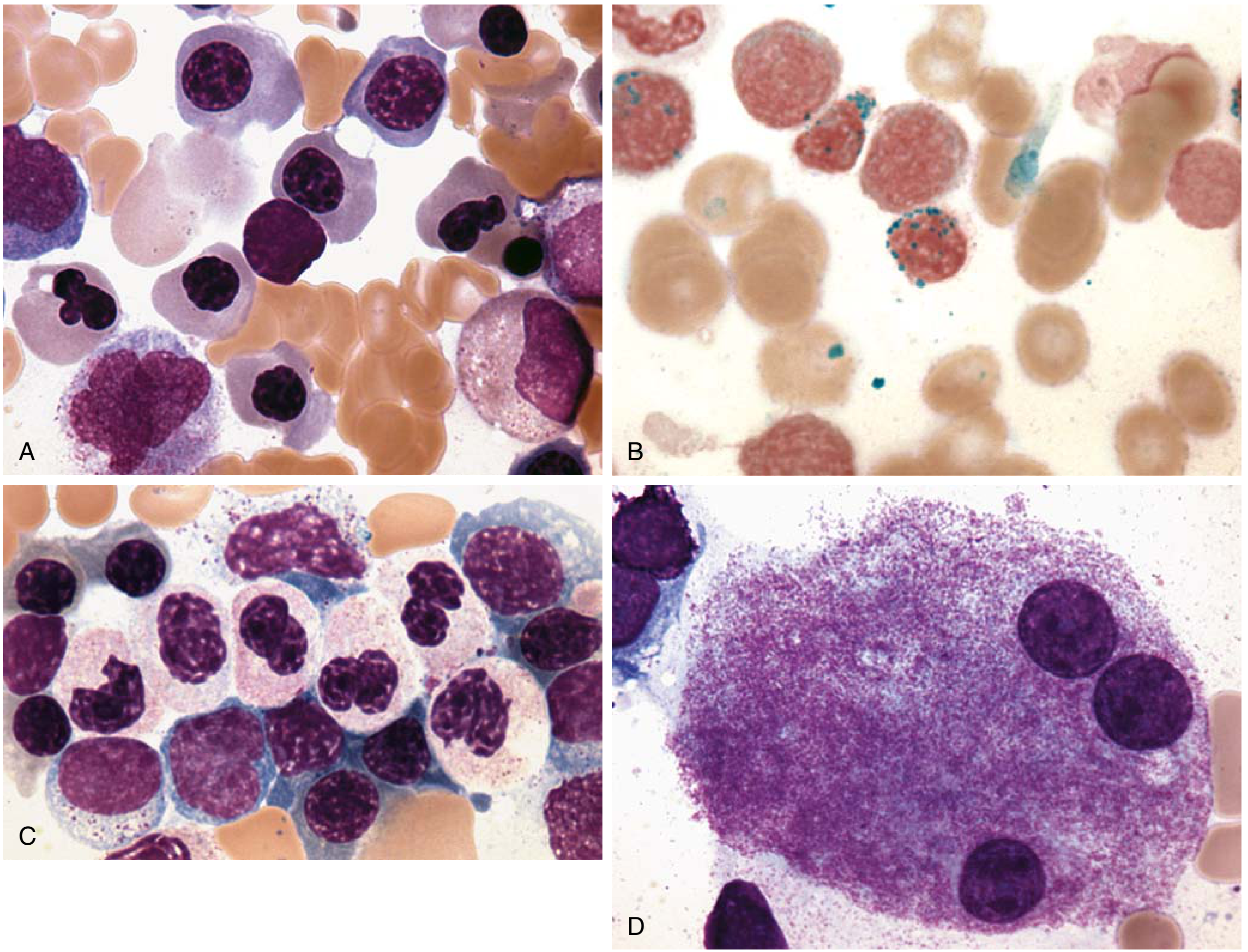
Fig: MDS bone marrow - (A) dyserythropoiesis, (B) ring sideroblasts (Prussian blue), (C) dysgranulopoiesis with pseudo-Pelger-Huët cells, (D) dysmegakaryopoiesis. (Source: Robbins Basic Pathology)
C. Cytogenetics and Molecular Studies
- Conventional karyotyping (G-banding): Standard; detects structural/numerical chromosomal abnormalities in ~40-70% of cases
- FISH (Fluorescence In Situ Hybridization): For del(5q), del(7q), trisomy 8 when karyotype fails
- Molecular mutation panel: SF3B1, SRSF2, TET2, DNMT3A, ASXL1, RUNX1, TP53, IDH1/2, EZH2 (increasingly required for accurate classification and prognosis)
- Next-generation sequencing (NGS): Preferred for comprehensive mutation profiling
D. Iron Studies
- Serum ferritin elevated (due to transfusions, iron loading from ineffective erythropoiesis)
- Prussian blue stain on BM aspirate to identify ring sideroblasts
- Transferrin saturation may be elevated
E. Other Investigations
- Serum erythropoietin (EPO) level: Important for treatment planning; low EPO (or inappropriately normal) predicts good response to ESA therapy
- Serum LDH: Often elevated (marker of ineffective hematopoiesis/cell turnover)
- Reticulocyte count: Inappropriately low (reticulocytopenia)
- Vitamin B12 and folate: To exclude megaloblastic anemia
- Flow cytometry: Immunophenotyping of aberrant hematopoietic populations; useful when morphology is equivocal
- Serum beta-2 microglobulin: Used in prognostic models
Prognostic Markers
IPSS (International Prognostic Scoring System) - Original 1997
The original IPSS uses three variables:
| Variable | 0 | 0.5 | 1.0 | 1.5 | 2.0 |
|---|---|---|---|---|---|
| % BM blasts | <5% | 5-10% | - | 11-20% | 21-30% |
| Cytogenetics | Good | Intermediate | Poor | - | - |
| Cytopenias | 0/1 | 2/3 | - | - | - |
- Good cytogenetics: Normal, -Y, del(5q), del(20q)
- Poor cytogenetics: Complex (≥3 abnormalities), chromosome 7 abnormalities
- Intermediate: All others
Risk groups:
- Low (0): Median survival ~5.7 years; 25% AML transformation at 9.4 years
- INT-1 (0.5-1.0): ~3.5 years; AML at 3.3 years
- INT-2 (1.5-2.0): ~1.2 years; AML at 1.1 years
- High (≥2.5): ~0.4 years; AML at 0.2 years
IPSS-R (Revised IPSS, 2012)
Improved system with 5 cytogenetic categories and refined blast thresholds:
| Cytogenetic Group | Examples |
|---|---|
| Very good | -Y, del(11q) |
| Good | Normal, del(5q), del(12p), del(20q), double inc. del(5q) |
| Intermediate | del(7q), +8, +19, i(17q), any other single or double |
| Poor | -7, inv(3)/t(3q)/del(3q), double inc. -7/del(7q), complex (3 abnormalities) |
| Very poor | Complex >3 abnormalities |
IPSS-R risk groups: Very Low, Low, Intermediate, High, Very High - with median survivals from >8.8 years down to 0.8 years.
IPSS-M (Molecular IPSS, 2022)
The newest prognostic model incorporates 31 gene mutations alongside cytogenetics and clinical variables. It identifies 6 risk groups and outperforms IPSS-R for predicting outcomes, especially identifying patients reclassified from "low risk" to higher risk based on mutations (e.g., RUNX1, TP53 biallelic, NRAS, PTPN11).
Additional Prognostic Markers
- TP53 mutation (especially biallelic): Worst prognosis regardless of other factors; associated with complex karyotype; often therapy-related
- RUNX1, ASXL1, EZH2 mutations: Associated with inferior overall survival
- SF3B1 mutation: Favorable - associated with ring sideroblasts and indolent course
- IDH1/IDH2 mutations: Targetable; associated with higher-risk disease
- Serum ferritin >1000 ng/mL: Adverse prognostic factor (iron overload from transfusions)
- Age, performance status, serum LDH, beta-2 microglobulin: Incorporated in WHO-based prognostic scoring systems (WPSS)
Key overall prognostic principle: blast count >5% elevates risk; >10% elevates further; complex chromosomal abnormalities or chromosome 7 abnormalities = high risk; del(5q) isolated = low risk. - Henry's, p. 737
Note on Current Concepts in MDS
1. Clonal Hematopoiesis of Indeterminate Potential (CHIP)
A recently recognized pre-MDS state where individuals carry somatic mutations in MDS-associated genes (TET2, DNMT3A, ASXL1) without cytopenias or dysplasia. CHIP is present in ~10% of people over 70 years and confers a 0.5-1% annual risk of progression to MDS/AML. It is also a risk factor for cardiovascular disease.
2. New Drug Approvals and Targeted Therapy
- Luspatercept (Reblozyl): A TGF-β ligand trap approved for transfusion-dependent anemia in lower-risk MDS with ring sideroblasts (MDS-RS). It promotes late-stage erythroid differentiation. The MEDALIST trial showed 38% transfusion independence. Now also approved for MDS with SF3B1 mutation.
- Imetelstat (Rytelo): A telomerase inhibitor approved in 2024 for transfusion-dependent anemia in low-/intermediate-risk MDS not responsive to ESAs - represents a novel mechanism of action.
- Azacitidine (5-azacytidine) and Decitabine: DNA hypomethylating agents (HMAs) - standard of care for higher-risk MDS. They "reprogram" aberrant MDS stem cells and improve differentiation. Azacitidine prolongs survival vs. conventional care (AZA-001 trial).
- Decitabine/cedazuridine (oral): Oral HMA formulation with equivalent PK to IV decitabine.
- Venetoclax + HMA combinations: Active area of investigation for higher-risk MDS.
- IDH1/IDH2 inhibitors (ivosidenib, enasidenib): Active in IDH-mutated MDS.
- Magrolimab (anti-CD47): Phagocytic checkpoint inhibitor in trials for TP53-mutated MDS.
3. Allogeneic Hematopoietic Cell Transplantation (allo-HCT)
The only potentially curative treatment for MDS. Best results in younger patients with early disease. Reduced-intensity conditioning (RIC) regimens have extended eligibility to older patients (up to 70s). A randomized trial showed busulfan-based RIC achieved at least 2-year relapse-free and overall survival comparable to myeloablative conditioning. - Goldman-Cecil Medicine, p. 1856
4. VEXAS Syndrome
A recently described (2020) inflammatory disorder caused by somatic UBA1 mutations in hematopoietic stem cells, presenting in older men with cytopenias, vacuolation of myeloid and erythroid precursors in the marrow, and systemic autoinflammatory features (relapsing polychondritis, Sweet syndrome). It mimics/overlaps with MDS and should be considered in older males with unexplained cytopenias.
5. Revised WHO 2022 Classification (WHO-HAEM5)
- Renamed from "Myelodysplastic Syndromes" to "Myelodysplastic Neoplasms" (MDN) to emphasize neoplastic nature
- MDS with SF3B1 mutation now a distinct category
- Biallelic TP53 mutated MDS now a separate high-risk entity
- Low-blast MDS categories revised with molecular integration
6. Supportive Care Updates
- Iron chelation (deferoxamine, deferasirox) recommended when serum ferritin >1000-2500 ng/mL to prevent end-organ damage in transfusion-dependent patients
- ESA (epoetin alfa, darbepoetin) remains first-line for lower-risk MDS with serum EPO <500 mU/mL and low transfusion burden
Recent evidence note: A 2025 review (PMID: 40056069) by Gangat & Tefferi highlights emerging pathogenetic mechanisms and new drugs for anemia in myelofibrosis and MDS, including the role of activin receptor pathway inhibition. A 2025 review (PMID: 39633555) in Haematologica covers current management of high-risk MDS.
PART 2: MYELOPROLIFERATIVE DISORDERS (Myeloproliferative Neoplasms - MPN)
Definition
Myeloproliferative neoplasms (MPNs) are clonal disorders of pluripotent hematopoietic stem cells characterized by constitutive activation of signaling pathways (especially tyrosine kinases), resulting in growth factor-independent proliferation of one or more myeloid cell lines (granulocytes, erythroid cells, megakaryocytes) with preserved differentiation. The key distinction from MDS is that in MPNs, maturation is near-normal and blood counts are typically elevated (not reduced, at least early on). - Robbins Basic Pathology, p. 405; Robbins Pathologic Basis of Disease, summary box
Classification (WHO 2016/2022)
The WHO classifies the following MPNs:
- Chronic Myeloid Leukemia (CML), BCR-ABL1 positive
- Polycythemia Vera (PV)
- Primary Myelofibrosis (PMF) - including prefibrotic/early PMF and overt fibrotic PMF
- Essential Thrombocythemia (ET)
- Chronic Neutrophilic Leukemia (CNL)
- Chronic Eosinophilic Leukemia, NOS (CEL-NOS)
- Mastocytosis (sometimes classified separately)
- MPN, Unclassifiable (MPN-U)
The four major entities are CML, PV, PMF, and ET. - Henry's, p. 735; Robbins Pathologic Basis of Disease, p. 405
Pathophysiology
Core Mechanism: Constitutive Tyrosine Kinase Activation
Normal hematopoietic growth factors (EPO, TPO, G-CSF) bind surface receptors and activate JAK-STAT signaling transiently. In MPNs, somatic mutations create constitutively active tyrosine kinases that bypass normal growth factor requirements, driving continuous proliferation and survival of marrow progenitors. Because differentiation is preserved, the result is overproduction of mature blood elements. - Robbins Pathologic Basis of Disease, p. 58
Specific Mutations by Disease
| Disorder | Key Mutation | Frequency | Mechanism |
|---|---|---|---|
| CML | BCR::ABL1 fusion (Philadelphia chromosome) | 100% | Constitutive ABL kinase activation |
| Polycythemia vera | JAK2 V617F (or JAK2 exon 12) | >95% | Constitutive JAK2 activation |
| Essential thrombocythemia | JAK2 V617F | 50-60% | Constitutive JAK2 activation |
| CALR mutations | 25-35% | Alternative MPL ligand activation | |
| MPL mutations | 5-10% | Constitutive MPL kinase activation | |
| Primary myelofibrosis | JAK2 V617F | 50-60% | Constitutive JAK2 activation |
| CALR mutations | 25-35% | Alternative MPL ligand activation | |
| MPL mutations | 5-10% | Constitutive MPL kinase activation | |
| Mastocytosis | KIT D816V | >90% | Constitutive KIT kinase activation |
Source: Robbins Pathologic Basis of Disease, Table 13.11, p. 583
BCR-ABL Pathway (CML)
The t(9;22)(q34;q11) translocation - the Philadelphia chromosome - creates the BCR::ABL1 fusion gene encoding a 210 kDa constitutively active BCR-ABL tyrosine kinase. This activates RAS/MAPK, PI3K/AKT, and JAK/STAT pathways, driving massive granulocytic proliferation from a pluripotent HSC. Present in >90% of CML; remaining cases involve cryptic rearrangements. - Robbins, p. 583
JAK2 Pathway (PV, ET, PMF)
JAK2 V617F (valine-to-phenylalanine substitution at residue 617) mimics erythropoietin/thrombopoietin receptor activation, constitutively activating STAT5 and downstream proliferative pathways. Despite being the same mutation, the resulting phenotype (PV vs. ET vs. PMF) depends on the hematopoietic cell of origin, mutation burden (VAF), co-mutations (ASXL1, SRSF2 in PMF), and individual host factors. - Robbins Pathologic Basis of Disease, p. 93
Marrow Fibrosis in PMF
In primary myelofibrosis, abnormal and dysplastic megakaryocytes release platelet-derived growth factor (PDGF), TGF-β, and basic FGF, stimulating non-clonal marrow fibroblast proliferation. This reactive fibrosis is the hallmark - it is not part of the neoplastic clone. As fibrosis progresses, marrow function is lost, forcing hematopoiesis to the spleen and liver (extramedullary hematopoiesis), causing massive splenomegaly and hepatomegaly. - Robbins Pathologic Basis of Disease, p. 59
Common Features of All MPNs
- Increased proliferative drive in bone marrow (hypercellularity)
- Variable extramedullary hematopoiesis (spleen, liver, lymph nodes) - hepatosplenomegaly
- Variable progression to a "spent phase" with marrow fibrosis and peripheral cytopenias
- Variable transformation to acute leukemia (blast crisis)
- Thrombotic and hemorrhagic complications (platelet dysfunction + hyperviscosity)
Individual Diseases - Clinical and Laboratory Features
1. Chronic Myeloid Leukemia (CML)
Clinical: Insidious onset; fatigue, weight loss, abdominal fullness (massive splenomegaly), night sweats. Rarely, first presentation as blast crisis.
Laboratory Features:
Peripheral blood:
- WBC markedly elevated (often 50,000-500,000/mm³), predominantly granulocytes
- Complete spectrum of granulocytic maturation: myeloblasts (<10%), promyelocytes, myelocytes (prominent peak), metamyelocytes, bands, mature neutrophils
- Basophilia (consistently present) and eosinophilia (almost always)
- Low Leukocyte Alkaline Phosphatase (LAP) score - key distinguishing feature from reactive leukocytosis
- Monocytosis (absolute increase)
- Mild anemia and thrombocytosis (early); thrombocytopenia (late)
Bone marrow:
- Markedly hypercellular; M:E ratio markedly increased (up to 20:1 or more)
- All stages of granulocytic maturation; megakaryocyte clusters
- No or minimal dysplasia in chronic phase
Molecular:
- BCR-ABL1 by PCR or FISH: Definitive diagnosis; also monitors treatment response (BCR-ABL transcript quantitation)
- Karyotype: t(9;22) Philadelphia chromosome in >90%
Disease phases:
- Chronic phase: <10% blasts; manageable
- Accelerated phase: 10-19% blasts, basophilia >20%, new cytogenetic clonal evolution, progressive splenomegaly
- Blast crisis: ≥20% blasts (myeloid in 70%, lymphoid in 30%) - acute leukemia behavior
2. Polycythemia Vera (PV)
Clinical: Plethora, cyanosis, pruritus (especially after hot bath - histamine from basophils), headache, hypertension. Thrombosis (DVT, MI, stroke, Budd-Chiari syndrome) in ~25%. Splenomegaly. Hyperuricemia and gout (5-10%).
Laboratory Features:
Peripheral blood:
- Hemoglobin >16.5 g/dL (males) or >16 g/dL (females); hematocrit often ≥55%
- Elevated total red cell mass
- Trilineage increase: Raised WBC (12,000-50,000/mm³) and platelets (>500,000/mm³) in most
- Giant and functionally abnormal platelets
- Platelet aggregation defects (most commonly decreased response to epinephrine)
Bone marrow:
- Hypercellular with panmyelosis (proliferation of erythroid, granulocytic, and megakaryocytic lineages)
- Pleomorphic, enlarged megakaryocytes; absent iron stores
Biochemistry/molecular:
- Serum EPO: Low (suppressed by increased red cell mass) - key diagnostic and differentiating feature
- JAK2 V617F mutation: >95% (or JAK2 exon 12 mutation)
- Elevated uric acid, LDH
- Increased B12 and B12-binding protein
- Low LAP score normal or elevated (variable)
WHO diagnostic criteria for PV:
- Major: (1) Hb/Hct above threshold OR elevated red cell volume; (2) Hypercellular BM with panmyelosis; (3) JAK2 V617F or exon 12 mutation
- Minor: (1) Low serum EPO level
3. Essential Thrombocythemia (ET)
Clinical: Often asymptomatic (50% found incidentally). Hemorrhage or thrombosis in ~50%. Erythromelalgia (burning pain/redness in extremities due to microvascular arteriolar occlusion by platelet thrombi). Mild splenomegaly in 50%.
Laboratory Features:
Peripheral blood:
- Sustained platelet count ≥450 x 10⁹/L (usually >1000 x 10⁹/L) with giant and morphologically abnormal forms, megakaryocyte fragments
- Normal or mild neutrophilic leukocytosis
- Hypochromic microcytic anemia (if chronic blood loss from mucosal bleeding)
- Platelet function: Decreased aggregation to epinephrine (most characteristic), ADP, and collagen
- Prolonged bleeding time (minority ~17%)
Bone marrow:
- Markedly increased and enlarged megakaryocytes with mature cytoplasm and multilobulated nuclei, tendency to cluster
- Normal or slightly hypercellular marrow; no significant granulocytic or erythroid hyperplasia
- Minimal or no fibrosis (reticulin normal or mildly increased)
Molecular:
- JAK2 V617F: 50-60%
- CALR mutation (type 1 or 2): 25-35%
- MPL mutation: 5-10%
- "Triple negative" (no JAK2/CALR/MPL): ~5%
WHO Criteria for ET:
- Platelets ≥450 x 10⁹/L (sustained)
- BM: Proliferation of mainly megakaryocytes (enlarged, mature); no increase in granulopoiesis or erythropoiesis
- Not meeting criteria for PV, PMF, CML, MDS, or other myeloid neoplasm
- Presence of JAK2, CALR, or MPL mutation (or clonal marker)
4. Primary Myelofibrosis (PMF)
Clinical: >60 years; progressive anemia; massive splenomegaly (extramedullary hematopoiesis); hepatomegaly; fatigue, weight loss, night sweats (constitutional symptoms); hyperuricemia; secondary gout. Bone pain. Radiography: patchy osteosclerosis in up to 50%.
Laboratory Features:
Peripheral blood:
- Moderate to severe normochromic normocytic anemia
- Leukoerythroblastic blood picture: Nucleated RBCs + immature granulocytes (myelocytes, metamyelocytes) in peripheral blood - hallmark
- Teardrop-shaped RBCs (dacryocytes): Pathognomonic of marrow fibrosis/infiltration
- WBC normal or mildly reduced initially; may be elevated early
- Platelet count normal or elevated early; thrombocytopenia supervenes with disease progression
- Atypical/giant platelets
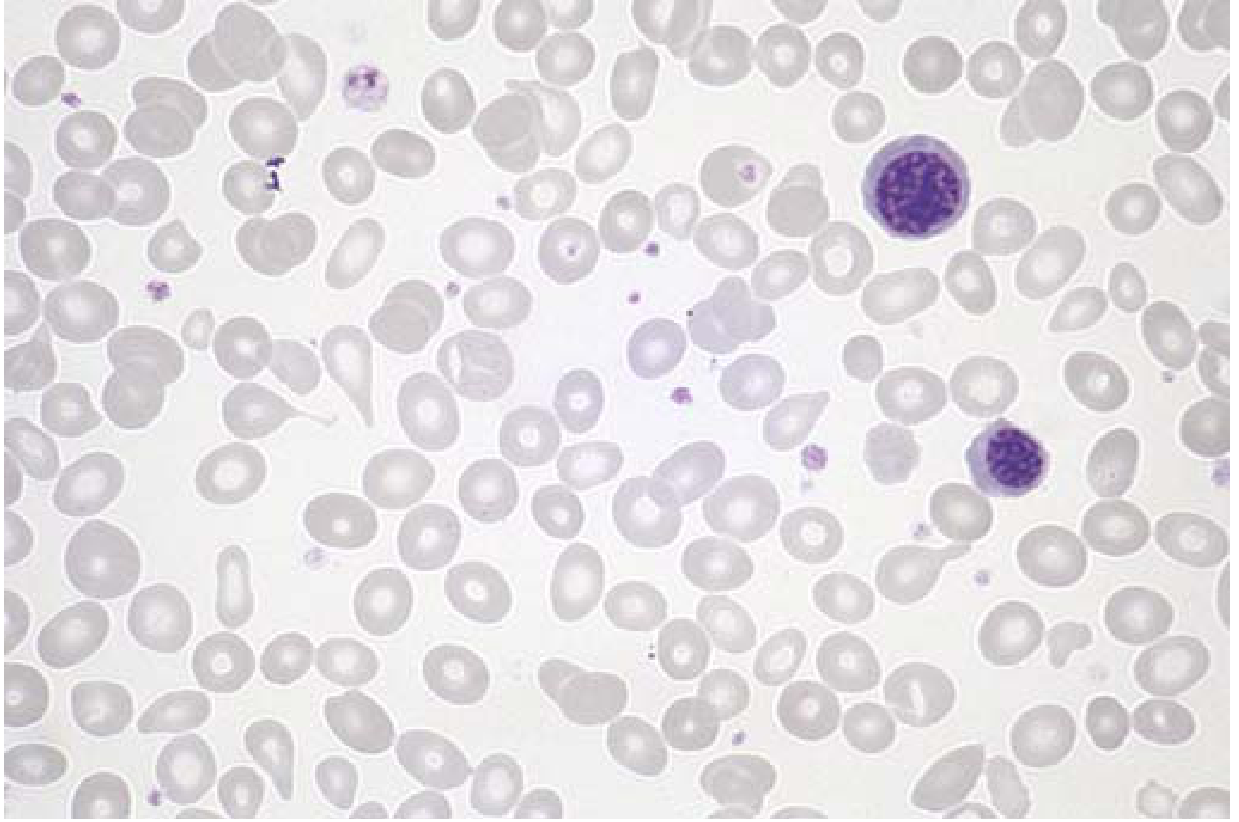
Fig: PMF peripheral blood smear - teardrop cells and nucleated erythroid precursors are visible (Source: Robbins Basic Pathology)
Bone marrow:
- Dry tap on aspiration (fibrosis)
- Trephine biopsy: Hypercellular early (prefibrotic stage); progressive reticulin/collagen fibrosis; atypical, enlarged megakaryocytes with hyperchromatic nuclei clustering around sinusoids and trabeculae; thickened bone trabeculae (osteosclerosis)
- The megakaryocyte hyperplasia with nuclear atypia + fibrosis is the hallmark
Molecular:
- JAK2 V617F: 50-60%; CALR: 25-35%; MPL: 5-10%
- Associated co-mutations (ASXL1, SRSF2, IDH1/2, EZH2) worsen prognosis
Prefibrotic PMF: 20-30% of cases; mild normocytic anemia, thrombocytosis, mild leukocytosis; hypercellular marrow with atypical megakaryocytes but without fibrosis - can mimic ET.
Summary Table of MPN Laboratory Findings
| Feature | CML | PV | ET | PMF |
|---|---|---|---|---|
| WBC | Very high | High | Normal/high | Normal/low |
| RBC/Hb | Normal/low | Markedly high | Normal/low | Low (anemia) |
| Platelets | Normal/high | High | Very high | Variable |
| LAP score | Low | High | Normal | Normal/high |
| Serum EPO | Normal | Low | Normal | Normal/low |
| Spleen | Markedly enlarged | Enlarged | Mildly enlarged | Massively enlarged |
| BM | Granulocytic hyperplasia | Panmyelosis | Megakaryocyte increase | Fibrosis + megakaryocyte atypia |
| Mutation | BCR-ABL1 | JAK2 >95% | JAK2/CALR/MPL | JAK2/CALR/MPL |
| Key blood finding | Full granulocytic spectrum, basophilia | Trilineage increase | Thrombocytosis | Leukoerythroblastosis, dacryocytes |
Differential Diagnosis with Supportive Laboratory Investigations
MPN vs. Reactive Conditions
Leukocytosis: CML vs. Leukemoid Reaction
| Feature | CML | Leukemoid Reaction |
|---|---|---|
| WBC | Usually >100,000 | <50,000 (usually) |
| Left shift | Myelocytes prominent | Less orderly; toxic granulation |
| Basophilia | Present (characteristic) | Absent |
| Eosinophilia | Present | Absent |
| LAP score | Low (<20) | High (>100) |
| Splenomegaly | Massive | Absent/mild |
| Philadelphia chromosome | Positive | Negative |
| BCR-ABL by PCR | Positive | Negative |
| Platelets | Elevated or normal | Normal |
| Anemia | Mild | Absent |
Key test: LAP score (leukocyte alkaline phosphatase) - low in CML, high in leukemoid reactions and PV. BCR-ABL PCR/FISH is definitive.
Erythrocytosis: PV vs. Secondary Polycythemia vs. Relative Polycythemia
| Feature | PV | Secondary Polycythemia | Relative Polycythemia |
|---|---|---|---|
| Red cell mass | Increased | Increased | Normal (plasma volume reduced) |
| WBC | Elevated | Normal | Normal |
| Platelets | Elevated | Normal | Normal |
| Splenomegaly | Present | Absent | Absent |
| Serum EPO | Low | High | Normal |
| JAK2 V617F | Positive (>95%) | Negative | Negative |
| BM | Panmyelosis | Erythroid hyperplasia only | Normal |
| SaO₂ | Normal | Low (if hypoxia cause) | Normal |
| Cause | Clonal | Hypoxia, EPO-secreting tumor | Stress, burns, dehydration |
Thrombocytosis: ET vs. Reactive Thrombocytosis
| Feature | ET | Reactive Thrombocytosis |
|---|---|---|
| Platelet count | >450, often >1000 | Usually <1000 x 10⁹/L |
| Platelet morphology | Giant, abnormal, megakaryocyte fragments | Normal or mildly large |
| Platelet function | Abnormal (decreased epinephrine aggregation) | Normal |
| Splenomegaly | Mild (50%) | Absent (or underlying cause) |
| Iron stores | Normal | Low iron (reactive thrombocytosis common with iron deficiency) |
| Serum ferritin/CRP | Normal | Elevated (iron deficiency or inflammation) |
| BM megakaryocytes | Increased, enlarged, multilobulated, clustered | Increased but normal morphology |
| JAK2/CALR/MPL | Positive in ~95% | Negative |
| Cause | Clonal | Infection, surgery, iron deficiency, splenectomy, malignancy |
PMF vs. Secondary Myelofibrosis
| Feature | PMF | Secondary Myelofibrosis |
|---|---|---|
| History | None | Prior MPN, malignancy, toxin exposure |
| BM fibrosis | Reticulin + collagen (progressive) | Variable |
| Megakaryocyte atypia | Marked | Less prominent |
| JAK2/CALR/MPL | Positive (>90%) | Negative (unless secondary to MPN) |
| Leukoerythroblastosis | Characteristic | May be present |
| Dacryocytes | Prominent | May be present |
| Splenomegaly | Massive | Variable |
| Causes (secondary) | - | Post-PV/ET, metastatic carcinoma, lymphoma, granuloma, radiation |
MPN vs. MPN Overlap
CML vs. CNL (Chronic Neutrophilic Leukemia)
| Feature | CML | CNL |
|---|---|---|
| WBC differential | Full spectrum (myelocytes prominent) | Predominantly mature neutrophils (>80%) |
| Basophilia | Characteristic | Absent |
| Eosinophilia | Present | Absent |
| BCR-ABL1 | Positive | Negative |
| CSF3R mutation | Negative | Positive (T618I) |
| Cytogenetics | t(9;22) | Normal (90%) or +8, +9 |
PV vs. PMF (Spent/Post-Polycythemic Phase)
In the spent phase of PV, myelofibrosis develops (~15-20% of cases after ~10 years), leading to anemia, massive splenomegaly, and leukoerythroblastosis - this can be indistinguishable morphologically from PMF. The key distinguishing feature is the clinical history of prior PV (documented erythrocytosis preceding the fibrotic phase). Both carry JAK2 V617F. Molecular co-mutation profiling may help.
ET vs. Prefibrotic PMF
This is one of the most diagnostically challenging distinctions:
| Feature | ET | Prefibrotic PMF |
|---|---|---|
| Anemia | Absent or mild | Mild to moderate |
| Leukoerythroblastosis | Absent | May be present |
| BM megakaryocyte morphology | Enlarged, mature, "staghorn" nuclei | Atypical, dense/hyperchromatic nuclei, cloud-like |
| BM fibrosis (reticulin) | Absent or grade 0-1 | Grade 0-1 but with atypical megakaryocytes |
| Splenomegaly | Mild | More prominent |
| Prognosis | Good | Worse (risk of overt fibrosis, AML) |
Requires BM trephine biopsy review by expert hematopathologist for definitive distinction.
Summary: Key Differential Tests at a Glance
| Investigation | What It Rules In / Rules Out |
|---|---|
| BCR-ABL1 PCR/FISH | Positive = CML; Negative = rules out CML in other MPN |
| JAK2 V617F | Positive in >95% PV, ~50% ET and PMF |
| CALR mutation | Positive in JAK2-negative ET and PMF; NOT in PV |
| MPL mutation | Positive in JAK2/CALR-negative ET and PMF |
| Serum EPO | Low in PV; high in secondary polycythemia |
| LAP score | Low in CML; high in leukemoid reaction and PV |
| BM trephine biopsy | Essential for PMF (fibrosis, megakaryocyte morphology) and for distinguishing ET vs. prefibrotic PMF |
| Platelet aggregation | Decreased to epinephrine in ET/PV |
| Red cell mass | Elevated in true polycythemia (PV and secondary); normal in relative polycythemia |
| CSF3R T618I | Diagnostic of CNL |
| Cytogenetics | t(9;22) in CML; complex = worse prognosis |
This is a shared conversation. Sign in to Orris to start your own chat.