in detail about gbs
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Guillain-Barre syndrome"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the information needed for a comprehensive answer. Here is the full detailed overview of Guillain-Barré Syndrome:
Guillain-Barré Syndrome (GBS) - A Detailed Overview
1. Definition
Guillain-Barré syndrome refers to a group of acquired, inflammatory polyradiculoneuropathies that share:
- Acute onset (maximum deficit by 4 weeks)
- Elevated CSF protein with low cell counts (cytoalbuminologic / albuminocytological dissociation)
- A monophasic course
First described in 1916 by Guillain, Barré, and Strohl, the key clinical features recognized were motor weakness, areflexia, paresthesias with minor sensory loss, and raised CSF protein without pleocytosis. With the eradication of poliomyelitis, GBS has become the leading cause of acute paralytic disease in Western countries.
- Goldman-Cecil Medicine, p. 4065
- Bradley and Daroff's Neurology in Clinical Practice, p. 2663
2. Epidemiology
| Parameter | Data |
|---|---|
| Annual incidence | 1-2 per 100,000 |
| AIDP incidence (Europe/North America) | 0.6-1.9 per 100,000 |
| Male:Female ratio | 1.4-1.5:1 |
| Age distribution | All ages; incidence rises with age (0.8 in <18 yrs, 3.2 in >60 yrs) |
| Preceding illness | 60% have prior respiratory or GI infection |
Notable epidemiological associations:
-
Campylobacter jejuni gastroenteritis is the most common preceding infection, especially in axonal variants
-
Zika virus is associated with a significantly increased risk of all GBS forms
-
Hepatitis E is a trigger in Belgium and Netherlands (5-10% of cases)
-
SARS-CoV-2 infection or most vaccines are NOT substantially associated with GBS (except a slight increase with ChADOx1nCoV-19 vaccine: 0.6 cases/100,000 doses)
-
GBS incidence actually declined during the COVID-19 pandemic due to reduced spread of usual infectious triggers
-
Goldman-Cecil Medicine, p. 4065-4066
3. Classification / Subtypes
Common Subtypes
| Subtype | Key Feature | Geography |
|---|---|---|
| AIDP (Acute Inflammatory Demyelinating Polyradiculoneuropathy) | Demyelination of nerve roots and peripheral nerves | 97% of cases in North America and Europe |
| AMAN (Acute Motor Axonal Neuropathy) | Pure motor; axonal injury; no sensory loss | Summer epidemics in China; children/young adults |
| AMSAN (Acute Motor Sensory Axonal Neuropathy) | Both motor and sensory axons; more severe; poor recovery | Less common |
Rare Variants
-
Miller-Fisher Syndrome (MFS): Classic triad of ophthalmoplegia, ataxia, and areflexia. Facial weakness, ptosis, and pupillary abnormalities may occur. NCS velocities are normal. Anti-GQ1b antibodies in >85% of cases. Accounts for 6% of GBS in Western countries, 18% in Taiwan.
-
Pharyngeal-cervical-brachial variant
-
Facial diplegia with paresthesias
-
Multiple cranial neuropathy variant
-
Paraparetic variant
-
Acute pandysautonomia
-
Bradley and Daroff's Neurology in Clinical Practice, p. 2663-2664
4. Pathobiology / Pathogenesis
All forms of GBS result from postinfectious molecular mimicry: the immune system attacks peripheral nerve antigens because they structurally resemble antigens presented by microbes, particularly C. jejuni.
In AIDP: Multifocal inflammatory demyelination of spinal roots and peripheral nerves, with mononuclear cell infiltrates rich in macrophages. Injury is most extensive in nerve roots and proximal nerve segments. Both T-cell-mediated and antibody-mediated responses are involved (T cells considered dominant).
In AMAN (shown in diagram below):
- The HS/0:19 serotype of C. jejuni shares GM1-like epitopes with axonal membranes
- This triggers production of anti-GM1 antibodies
- Activated complement components (C3d, C5b-9) and immunoglobulin deposit at nodes of Ranvier
- Macrophages are recruited to targeted nodes and invade the periaxonal space
- This leads to Wallerian degeneration
- Lack of blood-nerve barrier at motor nerve terminals makes distal axons especially vulnerable to circulating GM1 antibodies
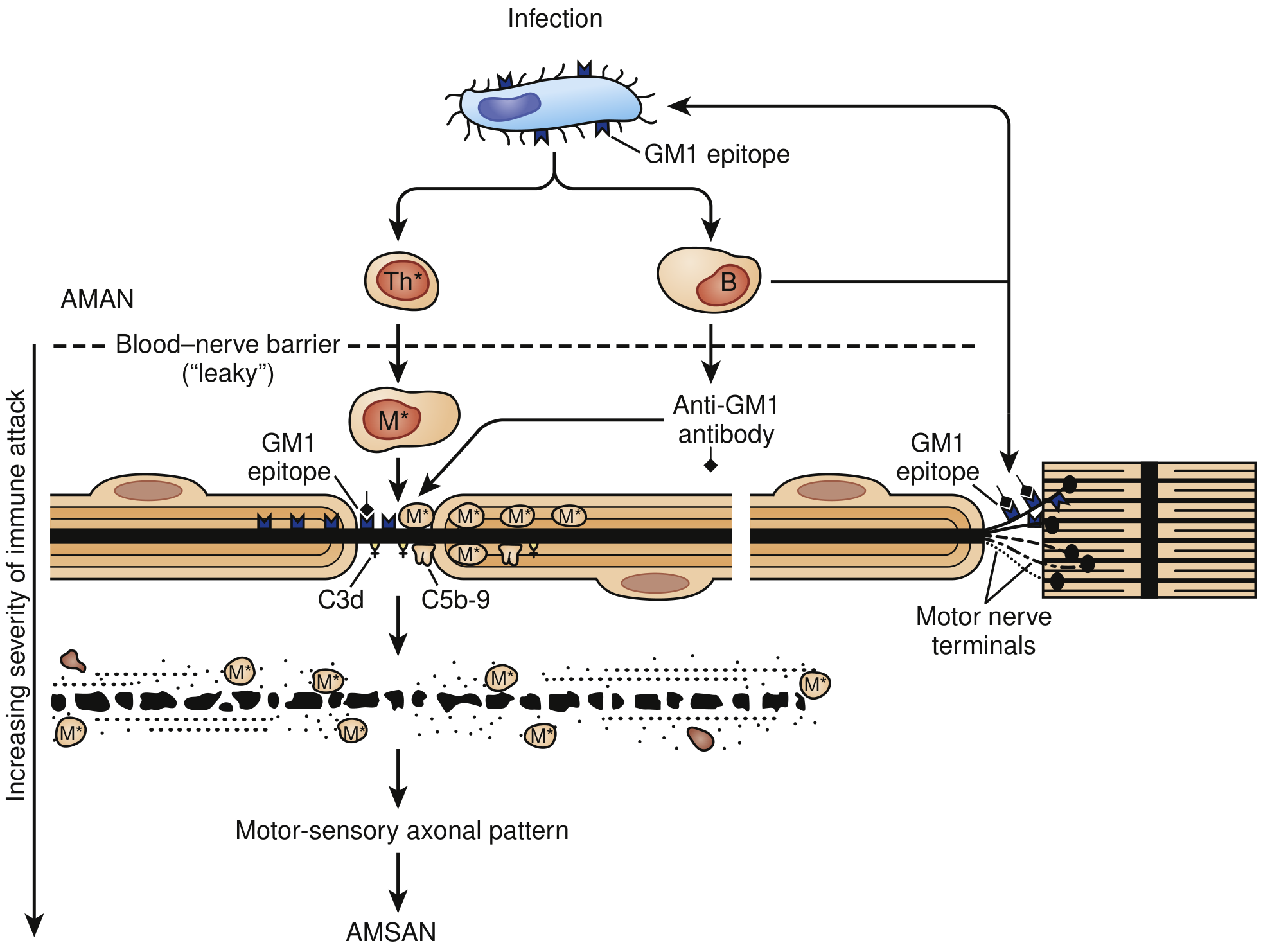
Fig: Immune Injury to Nerve Fibers in AMAN. Molecular mimicry of GM1-like epitopes between C. jejuni lipopolysaccharide and axonal membranes drives autoimmune attack. Increasing severity leads from AMAN to AMSAN. (Bradley and Daroff's Neurology, Fig. 106.18)
- Robbins & Kumar Basic Pathology, p. 809
- Goldman-Cecil Medicine, p. 4066
5. Clinical Features
Classic Presentation
- Weakness is the most common initial symptom - can range from mild difficulty walking to total quadriplegia with respiratory failure
- Classic ascending paralysis: leg weakness spreading to arms over hours to days
- Symmetrical weakness; proximal weakness is very common
- Areflexia/Hyporeflexia: invariable (may be absent early in disease)
- Symptoms progress over days to a maximum of 4 weeks (plateau then recovery)
Sensory Features
- Sensory loss is not prominent - often limited to distal vibration sense impairment
- Pain is underappreciated: moderate-to-severe pain in extremities, interscapular area, or back in ~70% during acute phase; may persist for a year in one-third
Cranial Nerve Involvement (45-75% of cases)
- Bilateral facial paresis in at least 50% of patients
- Extraocular muscle and lower cranial nerve involvement (less common)
- Occasional facial myokymia
- 5% present with isolated cranial nerve involvement that then descends
Autonomic Dysfunction (65% of cases)
- Cardiac arrhythmias (tachycardia, bradycardia)
- Blood pressure lability (hypertension and hypotension)
- Urinary retention
- Ileus
- Pupillary dysfunction
- Autonomic dysfunction is a major cause of mortality in ICU patients
Respiratory Involvement
-
Respiratory failure requiring mechanical ventilation in 9-30% of cases (increases with age)
-
Predictors of respiratory failure:
- Rapid disease progression (onset to admission <7 days)
- Severity of limb weakness
- Facial weakness
- Bulbar weakness
-
Goldman-Cecil Medicine, p. 4066
-
Bradley and Daroff's Neurology in Clinical Practice, p. 2663-2665
6. Diagnostic Criteria
Features Required for Diagnosis (Asbury & Cornblath criteria)
- Progressive weakness of both legs and arms
- Areflexia or hyporeflexia
Clinical Features Supportive of Diagnosis
- Progression over days to 4 weeks
- Relative symmetry
- Mild sensory symptoms or signs
- Bifacial palsies
- Autonomic dysfunction
- Absence of fever at onset
- Recovery beginning 2-4 weeks after progression ceases
Laboratory Features Supportive of Diagnosis
- Elevated CSF protein with <10 cells/µL (albuminocytologic dissociation)
- CSF protein may be normal in the first 7-10 days and remains normal in up to 10% of cases
- CSF WBC >50/mL suggests HIV or infection (Lyme disease)
- Electrodiagnostic features of nerve conduction slowing or block
- AIDP: reduced conduction velocities, conduction blocks
- AMAN/AMSAN: reduced compound muscle action potential (CMAP) amplitudes with relatively preserved conduction velocities
Antibodies
-
Anti-GQ1b: present in >85% of Miller-Fisher syndrome cases
-
Anti-GM1: present in AMAN, associated with C. jejuni infection
-
Bradley and Daroff's Neurology in Clinical Practice, Box 106.10, p. 2663
-
Goldman-Cecil Medicine, p. 4066-4067
7. Differential Diagnosis
Key conditions to exclude:
| Category | Conditions |
|---|---|
| Neuromuscular junction | Myasthenia gravis crisis, botulism, Lambert-Eaton |
| Spinal cord | Transverse myelitis, neuromyelitis optica, vascular myelopathy, cord compression |
| Anterior horn cell | Acute poliomyelitis, West Nile virus, enteroviruses |
| Toxic neuropathy | Arsenic, thallium, organophosphates, tick paralysis |
| Metabolic | Severe hypophosphatemia, hypermagnesemia, hypokalemia |
| Myopathy | Immune-mediated necrotizing myopathy (elevated CK), critical illness myopathy |
| Other neuropathies | Critical illness polyneuropathy, acute porphyria, diphtheria, vasculitic neuropathy |
Warning signs that suggest an alternative diagnosis:
-
Asymmetric weakness
-
Persistent bladder/bowel dysfunction
-
CSF cell count >50/mL
-
Fever at onset
-
Sensory level (suggests spinal cord)
-
Sharp sensorimotor level on examination
-
Goldman-Cecil Medicine, p. 4067
-
Bradley and Daroff's Neurology in Clinical Practice, p. 2666
8. Treatment
Supportive Care (Mainstay)
All GBS patients with worsening disease should be hospitalized. ICU admission is indicated for those with:
- Any predictors of respiratory failure
- Evidence of dysautonomia
- Signs of respiratory insufficiency
Respiratory Monitoring - "20-30-40 Rule":
- Vital capacity < 20 mL/kg (or 30% decline from baseline)
- Maximal inspiratory pressure < 30 cm H₂O
- Maximal expiratory pressure < 40 cm H₂O
Elective intubation should be performed when:
- FVC falls below 12-15 mL/kg (or <18 mL/kg with severe oropharyngeal weakness)
- Arterial PO₂ falls below 70 mmHg on room air
- Tracheostomy is required when ventilatory support is needed for >2 weeks
Monitor: pulse oximetry, heart rate, blood pressure, FVC every 4-6 hours when awake.
Immunotherapy - Two Equivalent First-Line Treatments
| Treatment | Dose | Mechanism | Notes |
|---|---|---|---|
| Intravenous Immunoglobulin (IVIG) | 0.4 g/kg/day x 5 days (total 2 g/kg) | Suppresses immune response (exact mechanism unclear) | Preferred for most patients; easier to administer |
| Plasmapheresis (plasma exchange) | 5 exchanges over 2 weeks | Removes offending antibodies from circulation | Equivalent to IVIG; not combined with IVIG |
Important notes:
- IVIG and plasmapheresis are equally effective - neither is clearly superior
- Corticosteroids are NOT effective in GBS and are not recommended
- Combining IVIG + plasmapheresis offers no additional benefit
- Treatment is most effective when started within 4 weeks of symptom onset
Supportive Measures
-
DVT prophylaxis (enoxaparin + compression stockings)
-
Pain management (gabapentin, carbamazepine for neuropathic pain; opioids for severe acute pain)
-
Urinary catheterization if needed
-
Bowel management for ileus
-
Cardiac monitoring for arrhythmia
-
Nutritional support (NG or PEG feeding in prolonged cases)
-
Rehabilitation (physiotherapy, occupational therapy)
-
Bradley and Daroff's Neurology in Clinical Practice, p. 2668-2670
-
Goldman-Cecil Medicine, p. 4067
-
Robbins & Kumar Basic Pathology, p. 809
9. Prognosis
| Timeframe | Data |
|---|---|
| Reach maximum disability by 2 weeks | 50% of patients |
| Reach maximum disability by 3 weeks | 75% of patients |
| Reach maximum disability by 4 weeks | >90% of patients |
| Mortality at 6 months (with modern care) | ~3% (was 33% before positive-pressure ventilation) |
| Full or near-full recovery | ~80% of patients |
| Persistent disability | ~20% |
Poor prognostic indicators:
- Older age
- Preceding diarrheal illness (especially C. jejuni)
- Axonal variants (AMAN, AMSAN)
- Severe weakness at nadir
- Reduced CMAP amplitudes in upper extremities (indicating axonal loss)
- Need for mechanical ventilation
After the acute phase, slow recovery occurs over weeks to months. Pain and fatigue can persist for a year or more in a significant minority.
- Goldman-Cecil Medicine, p. 4067
- Bradley and Daroff's Neurology in Clinical Practice, p. 2670
10. GBS vs. CIDP - Key Distinction
| Feature | GBS | CIDP |
|---|---|---|
| Time course | Acute - maximum deficit ≤4 weeks | Chronic - progresses >2 months; relapsing-remitting |
| Course | Monophasic | Chronic or relapsing |
| Treatment | IVIG or plasmapheresis (short course) | IVIG, plasmapheresis, or corticosteroids (long-term) |
| Steroids | Ineffective | Effective |
| Prognosis | Usually good recovery | Variable; may cause long-term disability |
Summary
GBS is an acute immune-mediated polyradiculoneuropathy triggered most commonly by Campylobacter jejuni infection through molecular mimicry. It presents as ascending weakness with areflexia, reaching maximum disability within 4 weeks. Respiratory failure (needing ventilation in ~30%) and autonomic instability are the life-threatening complications. Diagnosis rests on clinical features, CSF albuminocytologic dissociation, and nerve conduction studies. Treatment with IVIG or plasmapheresis is equally effective; steroids are contraindicated. With modern ICU care, mortality has fallen to ~3%, and ~80% of patients make a good recovery.
Recent evidence note: A 2026 systematic review (PMID: 41038424) confirms that physical exercise programs benefit fatigue and functional capacity in GBS and CIDP during rehabilitation - supporting active physiotherapy in recovery.
how to examine cns
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive textbook content from Harrison's and Bradley/Daroff's covering all components of the CNS examination. I have enough to write the complete detailed answer.
How to Examine the Central Nervous System (CNS)
The neurological examination is a systematic assessment of every region of the nervous system. It begins the moment you meet the patient - observing facial expression, posture, gait, and speech. Below is a structured, comprehensive guide based on standard clinical neurology practice.
Overview - Components of the CNS Examination
| Component | What It Tests |
|---|---|
| 1. Mental Status | Cortex, limbic system, frontal/temporal lobes |
| 2. Cranial Nerves I-XII | Brainstem, cranial nerve nuclei and pathways |
| 3. Motor System | Corticospinal tract, basal ganglia, LMN, muscle |
| 4. Coordination & Cerebellar | Cerebellum, spinocerebellar tracts |
| 5. Sensory System | Dorsal columns, spinothalamic tract, cortex |
| 6. Reflexes | UMN/LMN, spinal cord segments |
| 7. Gait & Balance | Integration of motor, sensory, cerebellar |
The experienced clinician uses a focused examination guided by the history, plus a screening examination of everything else. More complex functions are tested first - a patient who tandem walks normally does not have a significant cerebellar or proprioceptive problem.
- Bradley and Daroff's Neurology in Clinical Practice, p. 28
1. Mental Status Examination
Begin during history-taking - observe the patient's level of consciousness, attention, language, memory, and behaviour.
Consciousness & Alertness
- Note whether the patient is alert, confused, lethargic, or comatose
- Use the Glasgow Coma Scale (GCS) if impaired: Eye (1-4) + Verbal (1-5) + Motor (1-6)
Orientation
- Ask: person (name), place, date, year
Attention
- Ask to recite months backward, or serial 7s (subtract 7 from 100 repeatedly)
- Digit span forward (normal ≥6) and backward (normal ≥4)
Language (4 components)
- Fluency: Is speech fluent or non-fluent?
- Comprehension: Follow a 2-3 step command ("Pick up the paper with your right hand, fold it, and put it on the table")
- Repetition: Repeat a phrase ("No ifs, ands, or buts")
- Naming: Name common objects (watch, pen, knuckle)
- Note: Non-fluent aphasia + impaired comprehension = Broca's area lesion (left frontal); Fluent aphasia + impaired comprehension = Wernicke's area (left temporal)
Memory
- Immediate recall: Repeat 3 words
- Short-term: Recall 3 words after 5 minutes
- Long-term: Ask about past events
Visuospatial Function
- Ask to copy intersecting pentagons or draw a clock face
- Impaired in right parietal lobe lesions (constructional apraxia)
Higher Functions
- Praxis: Can patient mime brushing teeth or combing hair?
- Calculation: Serial 7s, simple arithmetic
- Abstraction: Interpret a proverb
- Judgment & insight
Quick Bedside Cognitive Tools
- Mini-Mental State Examination (MMSE): 30 points; <24 suggests cognitive impairment
- Montreal Cognitive Assessment (MoCA): More sensitive for mild cognitive impairment
Observational clues: hypomimia suggests parkinsonism; a worried/astonished expression may suggest progressive supranuclear palsy; ptosis may indicate myasthenia gravis or a brainstem lesion; the pattern of speech may reveal dysarthria or aphasia from the outset.
- Bradley and Daroff's Neurology, p. 28
2. Cranial Nerve Examination
Test cranial nerves in numerical order (group III, IV, VI together).
CN I - Olfactory Nerve
- Test when: spontaneous loss of smell, suspected Parkinson's disease, or head injury
- Method: With eyes closed, ask the patient to sniff a mild familiar odour (coffee, toothpaste) and identify it
- Anosmia: inferior frontal lobe lesion (e.g., meningioma), Parkinson's disease, head trauma
CN II - Optic Nerve
- Visual acuity: Test each eye separately using a Snellen chart (with glasses on)
- Visual fields by confrontation: Face the patient at ~60 cm; compare their fields to yours; move a finger in each quadrant (inferior then superior); test both eyes simultaneously first, then separately
- Bitemporal hemianopia: optic chiasm lesion (e.g., pituitary adenoma)
- Homonymous hemianopia: optic tract/radiation or occipital cortex lesion
- Pupillary light reflex / RAPD (Swinging flashlight test): Shine light alternately into each eye; relative afferent pupillary defect (RAPD) = optic nerve lesion on affected side
- Fundoscopy: Examine the optic disc (color, margins, cup-disc ratio), vessels, and retina
- Papilloedema: raised intracranial pressure
- Optic atrophy: previous optic neuritis or compression
CN III, IV, VI - Oculomotor, Trochlear, Abducens
- Pupil size and symmetry: Note anisocoria (unequal pupils)
- Large unreactive pupil: CN III palsy (compression, herniation)
- Small pupil + ptosis + anhidrosis: Horner syndrome (sympathetic pathway lesion)
- Pinpoint pupils: pontine lesion
- Accommodation reflex: Ask patient to follow finger moving to nose - pupil should constrict
- Extraocular movements (EOM): Ask patient to follow finger in an "H" pattern; look for:
- Paresis (limited movement)
- Diplopia in any direction (ask the patient directly)
- Nystagmus: fast and slow phases; note direction and type
- Horizontal nystagmus assessed at 45°, not at extreme gaze
- CN III palsy: Ptosis, "down and out" eye, dilated unreactive pupil
- CN IV palsy: Vertical diplopia; head tilt; hypertropia
- CN VI palsy: Esotropia; failure of abduction; horizontal diplopia
CN V - Trigeminal Nerve
- Sensory: Test light touch and pinprick in all three divisions (V1: forehead/scalp; V2: cheek/upper lip; V3: lower jaw/chin) on each side
- Corneal reflex: Lightly touch the cornea with a wisp of cotton (not conjunctiva); normal = blink bilaterally (afferent = CN V; efferent = CN VII)
- Motor (V3): Ask to clench teeth - palpate masseters; test jaw deviation against resistance (pterygoids)
- Jaw deviates toward side of lesion (weak pterygoid)
CN VII - Facial Nerve
- Inspect face at rest for asymmetry
- Test:
- Raise eyebrows (frontalis)
- Forcefully close eyes (orbicularis oculi) - try to open them
- Show teeth / smile (orbicularis oris)
- Puff out cheeks
- Key distinction:
- UMN (central) facial palsy: Lower two-thirds weakness only (forehead spared) - contralateral hemisphere/cortex lesion
- LMN (peripheral) facial palsy: Entire ipsilateral face weak including forehead - e.g., Bell's palsy, CN VII compression
CN VIII - Vestibulocochlear Nerve
- Hearing: Rub fingers near each ear; ask patient to detect whisper at ~60 cm
- Rinne test: Vibrating tuning fork (512 Hz) on mastoid until no longer heard, then at ear - normally air > bone (Rinne positive); if bone > air = conductive loss
- Weber test: Vibrating fork on forehead midline - normal lateralizes to neither side; lateralizes to bad ear in conductive loss, to good ear in sensorineural loss
- Vestibular function: Head impulse test, Dix-Hallpike maneuver for BPPV, nystagmus assessment
CN IX & X - Glossopharyngeal & Vagus
- Ask patient to say "Ahh" - palate should rise symmetrically in midline
- Unilateral palsy: uvula deviates away from lesion side
- Gag reflex: Touch posterior pharynx with tongue depressor (afferent CN IX; efferent CN X)
- Listen for hoarseness (recurrent laryngeal nerve, CN X)
- Assess swallowing
CN XI - Spinal Accessory Nerve
- Trapezius: Ask patient to shrug shoulders against resistance
- Sternocleidomastoid: Ask patient to turn head to the opposite side against resistance
CN XII - Hypoglossal Nerve
-
Ask patient to protrude tongue
-
Tongue deviates toward the side of an LMN lesion (weak side)
-
Look for fasciculations or wasting (LMN lesion)
-
Tongue deviates away from an UMN (contralateral cortical) lesion
-
Harrison's Principles of Internal Medicine 22E, p. 3423-3425
-
Goldman-Cecil Medicine, p. 464-480
3. Motor Examination
Step 1: Appearance (Inspection & Palpation)
- Look for muscle wasting/atrophy (LMN lesion, disuse)
- Look for fasciculations at rest (LMN - anterior horn cell disease, e.g., ALS)
- Look for involuntary movements:
- Resting tremor (Parkinson's disease - "pill-rolling")
- Postural tremor (essential tremor)
- Intention tremor (cerebellar disease)
- Chorea, athetosis, dystonia, myoclonus, tics
Step 2: Tone
Test resistance to passive movement with the patient relaxed (distract them if needed):
| Type of Tone Change | Character | Lesion |
|---|---|---|
| Spasticity | Velocity-dependent resistance; "clasp-knife" | UMN (corticospinal tract) |
| Rigidity | Uniform resistance in all directions; "lead pipe" | Extrapyramidal (basal ganglia) |
| Cogwheel rigidity | Ratchet-like jerky resistance | Parkinsonism |
| Paratonia | Fluctuating resistance | Frontal lobe or normal difficulty relaxing |
| Hypotonia / Flaccidity | Reduced resistance | LMN or peripheral nerve lesion, cerebellar |
- Upper limbs: rapid pronation/supination, wrist flexion/extension
- Lower limbs (supine): hands under knees, raise rapidly - normal heel drags; increased tone = heel lifts immediately
Step 3: Power (MRC Scale)
| Grade | Description |
|---|---|
| 0 | No movement |
| 1 | Flicker/trace contraction, no joint movement |
| 2 | Movement only with gravity eliminated |
| 3 | Movement against gravity, not against resistance |
| 4- | Movement against mild resistance |
| 4 | Movement against moderate resistance |
| 4+ | Movement against strong resistance |
| 5 | Full power |
UMN pattern of weakness: Extensors > flexors in upper limb; flexors > extensors in lower limb
LMN pattern: Distal weakness, wasting, fasciculations, hyporeflexia
Key screening tests for power:
-
Pronator drift: Both arms extended with eyes closed for 10 seconds - weak arm drifts downward and pronates (UMN lesion)
-
Upper limb: wrist/finger extensors (radial nerve, C7), grip, finger abduction
-
Lower limb: hip flexors, knee extensors, ankle dorsiflexion (L4/5), toe extension
-
Harrison's Principles of Internal Medicine 22E, p. 3424
4. Coordination & Cerebellar Examination
Limb Coordination
- Finger-to-nose test: Ask patient to touch their nose, then your finger, repeatedly; look for intention tremor and dysmetria (past-pointing)
- Heel-to-shin test: Ask patient to run one heel down the opposite shin repeatedly
- Rapid alternating movements (dysdiadochokinesis): Alternating pronation/supination of the hand rapidly; slowed, irregular = cerebellar dysfunction
Signs of Cerebellar Disease (DANISH mnemonic)
- Dysdiadochokinesis
- Ataxia (limb and gait)
- Nystagmus (horizontal, towards lesion)
- Intention tremor
- Scanning (slurred, staccato) speech
- Hypotonia
Note:
Limb incoordination from a cerebellar lesion will be associated with: ataxia, dysdiadochokinesis, nystagmus, and normal sensation.
If coordination is impaired with normal cerebellar signs but loss of proprioception, the cause is a sensory ataxia (dorsal column lesion).
5. Sensory Examination
Test primary modalities and higher cortical sensory functions.
Primary Modalities
| Modality | Pathway Tested | Method |
|---|---|---|
| Light touch | Dorsal columns (mainly) | Cotton wool on skin |
| Vibration | Dorsal columns | 128 Hz tuning fork on bony prominences (great toe, medial malleolus, tibial tuberosity) |
| Joint position sense (proprioception) | Dorsal columns | Hold toe/finger laterally; move up or down; patient identifies direction with eyes closed |
| Pain (pinprick) | Spinothalamic tract | Disposable pin; compare sides and proximal vs distal |
| Temperature | Spinothalamic tract | Hot and cold tubes on skin |
Testing strategy: Start distally; if abnormal, map the level proximally. Compare both sides.
Patterns of sensory loss:
- Stocking-glove distribution: Peripheral neuropathy (GBS, diabetes)
- Sensory level (below a dermatome): Spinal cord lesion
- Dissociated sensory loss (pain/temperature lost but vibration/proprioception intact): Anterior cord syndrome or syringomyelia
- Hemisensory loss: Contralateral hemisphere or thalamic lesion
- Crossed (face one side, body opposite): Brainstem lesion
Higher Cortical Sensory Functions (parietal lobe)
- Graphesthesia: Trace a number on the palm with eyes closed - patient identifies it
- Stereognosis: Patient identifies object placed in hand (coin, key) with eyes closed
- Two-point discrimination: Minimum distance to distinguish two simultaneous points
- Sensory extinction: Two areas touched simultaneously - parietal lesion causes extinction of one stimulus (usually contralateral)
6. Reflexes
Deep Tendon Reflexes (DTRs)
| Reflex | Level | Method |
|---|---|---|
| Biceps | C5, C6 | Tap biceps tendon at elbow |
| Brachioradialis | C5, C6 | Tap radius 5 cm above wrist |
| Triceps | C7, C8 | Tap triceps tendon above olecranon |
| Knee (patellar) | L3, L4 | Tap patellar tendon; knee jerk |
| Ankle (Achilles) | S1, S2 | Tap Achilles tendon; plantar flexion |
Grading:
- 0 = Absent
- 1+ = Diminished
- 2+ = Normal
- 3+ = Brisk (may be normal)
- 4+ = Clonus present (pathological; UMN lesion)
UMN lesion = Hyperreflexia, clonus, Babinski sign
LMN lesion = Hyporeflexia or areflexia
Plantar Response (Babinski Sign)
- Stroke the lateral sole with a blunt object from heel toward little toe, then curve medially
- Normal: Plantar flexion of toes (downgoing)
- Abnormal (Babinski positive / extensor): Big toe dorsiflexion ± fanning of other toes = UMN lesion above S1
- Present normally in infants up to 2 years (myelination not complete)
Superficial Reflexes
- Abdominal reflexes: Stroke each quadrant toward umbilicus; umbilicus moves toward stimulus; absent = UMN lesion ipsilaterally (T8-T12)
- Cremasteric reflex: Stroke inner thigh - testicle rises (L1, L2); absent = UMN or LMN lesion
- Anal reflex (S3, S4): Touch perianal skin; sphincter contracts
Primitive Reflexes (release signs = frontal lobe disease)
- Grasp reflex: Stroke patient's palm - fingers grasp examiner's fingers
- Palmomental reflex: Stroke thenar eminence - chin muscle twitches ipsilaterally
- Snout/pout reflex: Tap philtrum - pursing of lips
Special Reflexes
- Hoffman's sign: Flick terminal phalanx of middle finger; thumb and index flex = hyperreflexia (UMN lesion in cervical cord or above)
- Clonus: Rapidly dorsiflex the foot; rhythmic oscillations = UMN lesion
7. Gait and Balance
Observe gait spontaneously - note stance, base width, cadence, arm swing, and stride length.
| Gait Type | Characteristics | Lesion |
|---|---|---|
| Hemiparetic | One arm not swinging; leg circumducts | Contralateral hemisphere (stroke) |
| Spastic scissor | Legs cross over each other; stiff | Bilateral UMN (spinal cord or bilateral cortical) |
| Parkinsonian (festinant) | Shuffling, small steps, stooped, en bloc turns, reduced arm swing | Basal ganglia |
| Cerebellar (ataxic) | Wide base, irregular, staggering, worse eyes open AND closed | Cerebellum |
| Sensory ataxic | Wide-based, positive Romberg, worse eyes closed | Dorsal columns / peripheral neuropathy |
| Steppage | High-stepping; foot drop; slaps the ground | Common peroneal nerve or L4/5 |
| Waddling | Bilateral hip abductor weakness | Myopathy, bilateral hip disease |
| Antalgic | Reduced weight-bearing on one side | Pain |
Romberg Test
- Patient stands with feet together, arms at sides:
- Eyes open: stable
- Eyes closed: if the patient falls = Romberg positive
- Positive Romberg = loss of proprioception (dorsal column or peripheral sensory neuropathy) - the patient compensates visually; removing vision unmasks the deficit
- In cerebellar ataxia, the patient is unsteady with both eyes open and closed (Romberg is negative)
Tandem Gait (Heel-to-Toe Walking)
- Walk heel-to-toe along a straight line
- Fails with cerebellar midline disease or significant sensory loss
Postural Stability (Pull Test)
- Stand behind patient; pull back on shoulders briskly
- Normal: 1-2 corrective steps
- Abnormal (retropulsion): Parkinson's disease, PSP
8. Localisation Summary
Use findings to localise the lesion (essential step in neurological diagnosis):
| Level | Key Signs |
|---|---|
| Cortex/Hemisphere | Cognitive impairment, aphasia, seizures, contralateral hemiparesis, hemianopia, cortical sensory loss |
| Brainstem | Cranial nerve palsies + crossed motor/sensory signs (ipsilateral face, contralateral body) |
| Cerebellum | Ipsilateral limb ataxia, dysarthria, nystagmus, hypotonia |
| Spinal cord | Sensory level, UMN signs below level, sphincter dysfunction, spared head |
| Anterior horn / LMN | Flaccid weakness, wasting, fasciculations, areflexia |
| Peripheral nerve | Stocking-glove sensory loss, hyporeflexia |
| Neuromuscular junction | Fatigable weakness (myasthenia), spared sensation |
| Muscle | Proximal weakness, spared sensation, no fasciculations |
- Harrison's Principles of Internal Medicine 22E, Table 433-1
- Bradley and Daroff's Neurology in Clinical Practice, p. 28-30
Quick Reference - Screening Neurological Examination (Bradley & Daroff)
| Component | What to Test |
|---|---|
| Mental status | Assessed during history |
| CN I | Smell (when indicated) |
| CN II | Acuity, fields, RAPD, fundoscopy |
| CN III/IV/VI | Pupils, EOM, nystagmus |
| CN V | Facial sensation, corneal reflex, jaw power |
| CN VII | Eye closure, smile, forehead wrinkling |
| CN VIII | Whisper test, Rinne/Weber if indicated |
| CN IX/X | Palate elevation, gag |
| CN XI | Shoulder shrug |
| CN XII | Tongue protrusion |
| Motor | Involuntary movements, muscle bulk, tone, power (all groups), pronator drift |
| Coordination | Finger-nose, heel-shin, rapid alternating movements |
| Reflexes | Biceps, triceps, brachioradialis, knee, ankle, plantars |
| Sensation | Pinprick/light touch on hands and feet, joint position in hallux and index finger, vibration at ankle |
| Gait & Balance | Spontaneous gait, tandem walk, Romberg test, pull test |
This structured approach ensures every part of the CNS is assessed, abnormalities are localised accurately, and appropriate investigations are ordered.
examine
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have all the detail needed. Here is the full clinical examination of a GBS patient:
Clinical Examination of a GBS Patient
Examining a patient with suspected or confirmed GBS is a structured, head-to-toe neurological assessment with special attention to the hallmark findings: ascending weakness, areflexia, autonomic instability, and respiratory compromise. The examination also monitors for progression and complications.
Step 1: General Inspection (Before Touching the Patient)
As soon as you enter the room, observe:
| What to Look For | Significance in GBS |
|---|---|
| Facial expression | Bilateral facial weakness (bilateral CN VII palsy in ~50%) - flat, expressionless face |
| Posture | Inability to hold upright posture; proximal weakness |
| Breathing pattern | Shallow, laboured breathing; use of accessory muscles = impending respiratory failure |
| Limb position | Flaccid limbs lying flat; no spontaneous movement |
| Nasogastric/PEG tube | Suggests bulbar involvement with dysphagia |
| Tracheostomy/ventilator | Severe disease with respiratory failure |
| Cardiac monitor | Autonomic instability being monitored |
Ask the patient about: preceding illness (fever, diarrhoea, URTI 1-4 weeks ago), time of symptom onset, rate of progression, and any breathing difficulty or swallowing problems.
Step 2: Vital Signs - CRITICAL in GBS
Monitoring vitals is not routine - it is life-saving in GBS:
| Parameter | What to Assess | Significance |
|---|---|---|
| Respiratory rate | Count over 1 minute | Tachypnoea = early respiratory failure |
| SpO₂ | Pulse oximetry | Desaturation = respiratory compromise |
| Forced Vital Capacity (FVC) | Spirometry at bedside | <20 mL/kg = critical; monitor every 4-6 hours |
| NIF (Negative Inspiratory Force) | Bedside manometer | <-30 cmH₂O = impending failure ("20-30-40 rule") |
| Blood pressure | Lying and standing | Wide swings (hypo + hypertension) = autonomic dysfunction |
| Heart rate | Rate and rhythm | Tachycardia/bradycardia/arrhythmias = autonomic dysfunction |
| Temperature | Look for infection | Fever at onset should make you reconsider the diagnosis |
The "20-30-40 Rule" for intubation risk:
- Vital capacity < 20 mL/kg
- Maximum inspiratory pressure < 30 cmH₂O
- Maximum expiratory pressure < 40 cmH₂O
If all 3 are met → immediate ICU transfer and elective intubation.
Step 3: Motor Examination - The Core of GBS
3a. Inspect
- Look for muscle wasting (usually absent early; may appear after prolonged disease)
- Look for fasciculations (rare in GBS; their presence suggests anterior horn cell disease - reconsider diagnosis)
- Note posture of limbs: flaccid, lying flat
3b. Tone
- Hypotonia / Flaccidity in all affected limbs - the hallmark of a lower motor neuron/peripheral nerve lesion
- In GBS: tone is reduced or absent (not spastic)
- Test by passive wrist flexion/extension, knee lift with heel drag
3c. Power - Test Systematically Using MRC Scale (0-5)
Test each muscle group bilaterally and note asymmetry:
Upper Limbs:
| Muscle Group | Nerve | Root |
|---|---|---|
| Shoulder abduction (deltoid) | Axillary | C5 |
| Elbow flexion (biceps) | Musculocutaneous | C5-C6 |
| Elbow extension (triceps) | Radial | C7 |
| Wrist extension | Radial | C6-C7 |
| Finger extension | Radial (posterior interosseous) | C7 |
| Grip / finger flexion | Median/Ulnar | C8 |
| Finger abduction | Ulnar | T1 |
Lower Limbs:
| Muscle Group | Nerve | Root |
|---|---|---|
| Hip flexion (iliopsoas) | Femoral | L2-L3 |
| Knee extension (quadriceps) | Femoral | L3-L4 |
| Knee flexion (hamstrings) | Sciatic | L5-S1 |
| Ankle dorsiflexion (tibialis anterior) | Deep peroneal | L4-L5 |
| Ankle plantarflexion (gastrocnemius) | Tibial | S1 |
| Great toe extension | Deep peroneal | L5 |
Pattern in GBS:
- Typically symmetrical
- Proximal and distal weakness (unlike polyneuropathy which is usually distal-predominant)
- Classically ascending: legs > arms > respiratory/bulbar muscles
- In severe disease: near-total quadriplegia
Pronator Drift Test: Arms extended in supination with eyes closed - weak arm drifts and pronates. In GBS this may be negative or abnormal due to flaccidity rather than UMN drift.
Head lift test: Ask the patient to lift their head off the pillow - inability to do so is a predictor of needing mechanical ventilation.
Step 4: Reflexes - The Most Consistent Finding
Deep Tendon Reflexes (DTRs):
| Reflex | Normal | In GBS |
|---|---|---|
| Biceps (C5-C6) | 2+ | Absent or diminished |
| Brachioradialis (C5-C6) | 2+ | Absent or diminished |
| Triceps (C7) | 2+ | Absent or diminished |
| Knee/Patellar (L3-L4) | 2+ | Absent - most consistent finding |
| Ankle/Achilles (S1) | 2+ | Absent - most consistent finding |
Areflexia or hyporeflexia is the invariable feature of GBS - though it may be absent very early in the disease course. If reflexes are brisk or hyperreflexia is present, seriously question the diagnosis.
Plantar Response (Babinski):
- In GBS: typically flexor (normal/downgoing) - as expected with an LMN/peripheral nerve lesion
- An extensor plantar (Babinski positive) in a suspected GBS patient suggests a concurrent UMN (spinal cord/central) lesion - investigate urgently
Step 5: Sensory Examination
Sensory loss in GBS is not prominent but present in most patients - it is motor-predominant.
What to Test:
| Modality | Method | Finding in GBS |
|---|---|---|
| Pinprick | Disposable pin, compare distally to proximally | Mild distal loss; glove-and-stocking pattern |
| Light touch | Cotton wool | Mild distal impairment |
| Vibration | 128 Hz tuning fork at great toe, medial malleolus | Distally reduced - most common sensory finding |
| Joint position sense (proprioception) | Move great toe/finger up or down | Usually relatively preserved in early GBS |
| Temperature | Cold/warm tubes | May be mildly impaired distally |
Sensory pattern in GBS:
- Distal > proximal (glove-and-stocking)
- Mild to moderate - motor deficits dominate
- Vibration sense is the most commonly affected modality
- Pain/paresthesias (burning, tingling) present in ~70% of patients - often severe
Pain Assessment:
- Ask specifically about back pain (interscapular/lumbar - a common early complaint in GBS, sometimes misdiagnosed as musculoskeletal)
- Dysesthetic pain: burning, tingling of limbs
- Deep aching limb pain (muscle/joint pain)
Important: if there is a sensory level on the trunk (sensation normal above a dermatome, absent below), this suggests a spinal cord lesion, NOT GBS - arrange urgent MRI spine.
Step 6: Cranial Nerve Examination
Cranial nerves are affected in 45-75% of GBS patients:
| CN | Test | Expected Finding in GBS |
|---|---|---|
| CN II | Visual acuity, visual fields, fundoscopy | Usually normal; papilloedema if ICP raised (rare) |
| CN III/IV/VI | Extraocular movements, pupil size | Ophthalmoplegia (in Miller-Fisher variant); check for ptosis |
| CN V | Facial sensation, corneal reflex | May be affected; corneal reflex loss |
| CN VII (KEY) | Raise brows, close eyes, show teeth | Bilateral facial weakness in ~50% - symmetric "facial diplegia" |
| CN IX/X | Palate elevation, gag reflex, voice | Bulbar weakness: nasal voice, dysphagia, impaired gag reflex |
| CN XI | Shoulder shrug | Neck flexor/extensor weakness |
| CN XII | Tongue protrusion | Tongue weakness (less common) |
Miller-Fisher Variant triad (if suspected):
- Ophthalmoplegia - eyes don't move fully
- Ataxia - unsteady gait/limbs
- Areflexia - absent reflexes (often without limb weakness)
Step 7: Coordination
- Finger-nose and heel-shin tests
- In classic GBS: incoordination is mainly due to weakness, not true cerebellar dysfunction
- In Miller-Fisher variant: true cerebellar-type ataxia is present
- Differentiate sensory ataxia (proprioception loss) from cerebellar ataxia using Romberg test
Step 8: Gait Assessment
Observe the patient walking (if they can):
| Gait Finding | Interpretation |
|---|---|
| Wide-based, unsteady | Sensory ataxia (proprioceptive loss) or cerebellar (MFS variant) |
| Steppage gait | Foot drop from distal weakness |
| Unable to walk at all | Moderate-severe disease |
| Unable to rise from chair without arms | Proximal lower limb weakness |
Functional grading (Hughes disability scale) - assess at each visit:
| Grade | Status |
|---|---|
| 0 | Healthy |
| 1 | Minor symptoms; runs normally |
| 2 | Walks 5 m unaided |
| 3 | Walks 5 m with aid |
| 4 | Bedridden/wheelchair |
| 5 | Requires ventilation |
| 6 | Death |
Step 9: Autonomic Assessment
Autonomic dysfunction in 65% of GBS patients - a major cause of mortality:
| System | What to Assess | Findings |
|---|---|---|
| Cardiovascular | HR and rhythm; BP lying/standing | Tachycardia/bradycardia; postural hypotension; hypertension |
| Bowel | Bowel sounds; last bowel movement | Ileus; constipation |
| Bladder | Urinary retention; catheter needed? | Urinary retention (neurogenic bladder) |
| Pupils | Size, reactivity | Iridoplegia (dilated unreactive pupils) |
| Sweating | Skin | Anhidrosis or episodic diaphoresis; acral vasoconstriction |
"Vagal spells": sudden bradycardia, heart block, or asystole triggered by tracheal suction or straining - watch for these in ICU patients. ECG changes (T-wave, ST, QT prolongation) can occur.
Step 10: Look for Complications
| Complication | Clinical Sign |
|---|---|
| Respiratory failure | Tachypnoea, accessory muscle use, SpO₂ drop, low FVC |
| DVT/PE | Calf swelling, tachycardia, pleuritic chest pain |
| Pneumonia/aspiration | Fever, crepitations on chest auscultation |
| Pressure sores | Inspect skin over bony prominences |
| Pain | Assess severity (VAS); often undertreated |
| Depression/anxiety | Mental status; mood |
| Urinary infection | Suprapubic tenderness, fever (from catheterization) |
Summary of Key Examination Findings in GBS
| Domain | Classic GBS Finding |
|---|---|
| Tone | Flaccid (hypotonia) |
| Power | Symmetric weakness, ascending, proximal + distal |
| Reflexes | Areflexia (most consistent sign) |
| Plantar response | Flexor (downgoing - LMN pattern) |
| Sensation | Mild distal loss; reduced vibration; glove-stocking |
| Cranial nerves | Bilateral facial weakness; bulbar; ophthalmoplegia (MFS) |
| Autonomic | BP/HR instability, urinary retention, ileus |
| Coordination | Impaired due to weakness (not cerebellar); ataxia in MFS |
| Gait | Weak, steppage, or unable to walk |
| Respiratory | FVC, NIF - monitor closely for failure |
Red Flags That Suggest Another Diagnosis (NOT GBS)
| Finding | Alternative Diagnosis to Consider |
|---|---|
| Fever at onset | Meningitis, encephalitis, Lyme, HIV |
| Brisk reflexes / Babinski +ve | Spinal cord lesion (myelopathy) |
| Sensory level | Transverse myelitis, cord compression |
| Bowel/bladder symptoms very early | Spinal cord disease |
| Asymmetric weakness | Stroke, mononeuritis, compressive lesion |
| CSF pleocytosis (>50 WBC) | HIV seroconversion, Lyme disease, CMV |
| Normal NCS early, ascending course | Consider polio, botulism, tick paralysis |
| Ophthalmoplegia + unreactive pupils | Botulism (no sensory loss, dry mouth) |
- Bradley and Daroff's Neurology in Clinical Practice, p. 2663-2670
- Neuroanatomy through Clinical Cases 3rd Edition, p. 407
- Goldman-Cecil Medicine, p. 4066-4067
anything more about gbs
Reading File
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
"Guillain-Barre syndrome"[MeSH Terms] AND diagnosis
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive additional detail on GBS. Here is a focused deep-dive covering areas not fully addressed before:
Additional GBS - Deeper Details
1. Electrodiagnostic Studies (NCS / EMG) - The Cornerstone of Subtype Classification
Nerve Conduction Studies (NCS) and EMG are essential in GBS - they confirm the diagnosis, classify the subtype (demyelinating vs axonal), and help predict prognosis.
AIDP (Demyelinating) - NCS Findings
| Parameter | Finding |
|---|---|
| Motor conduction velocity | Reduced (<70-80% of lower limit of normal) |
| Distal motor latency | Prolonged |
| F-wave latency | Prolonged or absent (early sign of root demyelination) |
| Compound Motor Action Potential (CMAP) | Reduced amplitude or conduction block |
| Sensory Nerve Action Potential (SNAP) | Abnormal (reduced or absent) |
| Conduction block | Present (hallmark of AIDP) |
| H-reflex | Absent (early, even before other changes) |
Conduction block = CMAP amplitude drops >50% from distal to proximal stimulation - indicates focal demyelination. This is what causes clinical weakness disproportionate to axonal damage.
AMAN (Axonal Motor) - NCS Findings
| Parameter | Finding |
|---|---|
| Motor conduction velocity | Normal |
| Distal motor latency | Normal |
| CMAP amplitude | Reduced or absent (axonal loss) |
| SNAP | Normal (pure motor - sensory spared) |
| Conduction block | Usually absent |
| Needle EMG | Fibrillations and positive sharp waves (denervation) appear weeks later |
AMSAN (Axonal Motor + Sensory) - NCS Findings
- Same as AMAN but SNAPs also reduced or absent
- Needle EMG: abundant fibrillations; persistently inexcitable motor nerves
- Worst prognosis of all subtypes
Miller-Fisher Syndrome (MFS) - NCS Findings
- SNAPs reduced or absent (sural sparing pattern in 1/3)
- Motor NCS usually normal
- F-waves and needle EMG usually normal
- CSF: elevated protein in ~70% at 1 week
Important pitfall: NCS may be normal in the first 7-10 days of GBS. If the clinical picture is strongly suggestive but NCS is normal, repeat in 1 week (see management flowchart below).
- Bradley and Daroff's Neurology, p. 2666-2670
2. Serological / Antibody Testing
| Antibody | Subtype / Variant | Clinical Value |
|---|---|---|
| Anti-GQ1b (IgG) | Miller-Fisher syndrome, Bickerstaff brainstem encephalitis | Present in 95-98% of MFS - highly specific |
| Anti-GM1 (IgG) | AMAN | Associated with C. jejuni; predicts poor prognosis |
| Anti-GD1a (IgG) | AMAN | Associated with C. jejuni |
| Anti-GD1b | AMSAN, sensory GBS | Sensory nerve involvement |
| Anti-GalNAc-GD1a | AMAN (China) | Motor-specific |
| Anti-ganglioside antibodies (AIDP) | None specific | NOT clinically useful in AIDP |
| Campylobacter serology | AMAN, AIDP | Identifies trigger; predicts axonal risk |
| CMV/EBV/VZV/Mycoplasma serology | AIDP | Identifies trigger only |
Ganglioside antibody testing is only clinically useful for MFS (anti-GQ1b) and axonal variants (anti-GM1). Testing is not recommended routinely in classic AIDP.
3. CSF Analysis - The "Albuminocytologic Dissociation"
| CSF Parameter | Classic Finding in GBS | Notes |
|---|---|---|
| Protein | Elevated (>45 mg/dL; often 100-1000 mg/dL) | Rises due to radiculitis and inflammation |
| Cell count (WBC) | <10 cells/µL - near normal | KEY: high protein with low cells = dissociation |
| Glucose | Normal | |
| Opening pressure | Normal | |
| Appearance | Clear |
Timing caveat: CSF protein may be normal in the first 7-10 days (remains normal in up to 10% of all cases). In early GBS with normal CSF, repeat LP after 1 week if suspicion is high.
Red flag - when to question the diagnosis:
- WBC > 50/µL → consider HIV seroconversion, Lyme disease, CMV radiculitis, lymphomatous meningitis
- WBC > 10 but < 50 may occasionally occur in GBS with concurrent HIV or CMV
4. MRI Findings
- Brain MRI: usually normal in classic GBS
- MRI spine with gadolinium: may show nerve root enhancement of the cauda equina - particularly in children. This reflects inflammation at the roots.
- MRI is primarily used to exclude alternative diagnoses (myelopathy, cord compression, brainstem lesion)
- In MFS: MRI brain occasionally shows brainstem lesions or gadolinium enhancement of ocular motor nerves
5. Subtype Deep-Dive
AMAN - Acute Motor Axonal Neuropathy
- First reported in epidemic proportions in children/young adults in northern China (summer months)
- Now the most common GBS subtype in Asia
- Pure motor: no sensory involvement clinically or electrophysiologically
- Normal SNAPs; reduced CMAP; no conduction slowing
- Pathology: macrophage intrusion between axon and myelin at nodes of Ranvier, leading to membrane-attack complex formation → conduction failure
- Anti-GM1 and anti-GD1a IgG antibodies (molecular mimicry with C. jejuni lipopolysaccharide)
- Paradox: most patients improve as rapidly as AIDP despite axonal pattern - because conduction failure is often reversible without true axonal degeneration
AMSAN - Acute Motor-Sensory Axonal Neuropathy
- Fulminant, rare, and severe
- Maximum deficit within <7 days from onset
- Profound quadriparesis, severe muscle wasting, prolonged ventilation
- Absent CMAPs + absent SNAPs; no conduction slowing
- Abundant fibrillations on needle EMG
- Extensive wallerian degeneration in ventral AND dorsal roots
- Poor prognosis - recovery is delayed and often incomplete
Miller-Fisher Syndrome (MFS)
- Classic triad: Ophthalmoplegia + Ataxia + Areflexia
- Diplopia is often the first symptom
- Ocular signs: from complete ophthalmoplegia (dilated unreactive pupils) to partial external ophthalmoparesis ± ptosis
- Ataxia: due to peripheral mismatch between proprioceptive input from muscle spindles and kinesthetic information from joint receptors
- Motor strength: characteristically preserved - but ~5% overlap with classic GBS and develop quadriparesis
- Anti-GQ1b IgG: 95-98% positive (most sensitive and specific test in neurology)
- Precipitants: C. jejuni (20%), H. influenzae (8%)
- Prognosis: excellent - most recover fully within weeks to months
- Related condition: Bickerstaff Brainstem Encephalitis (BBE) - also anti-GQ1b positive, presents with ophthalmoplegia + ataxia + altered consciousness (CNS involvement)
6. Management Flowchart
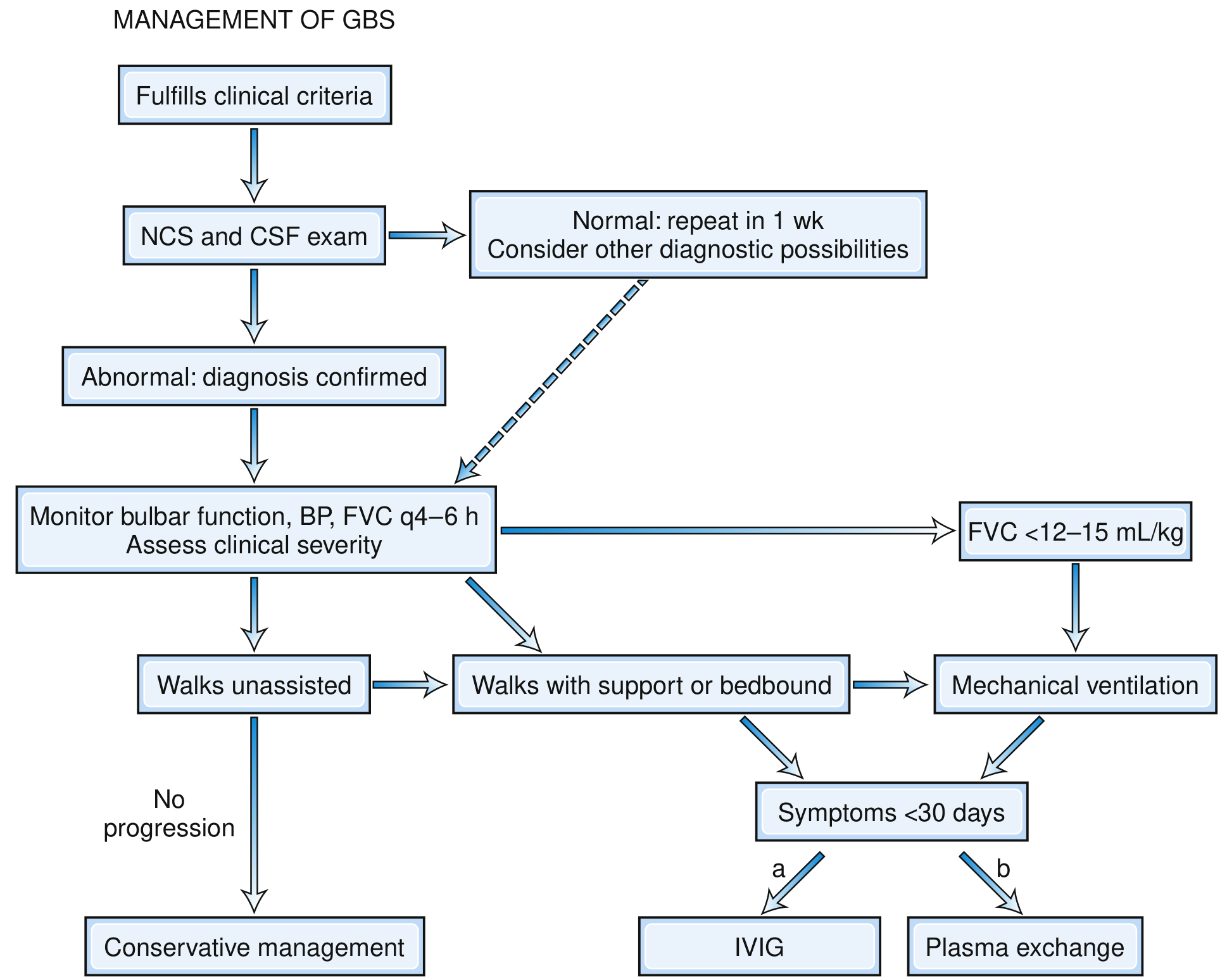
Fig. GBS Management Pathway (Bradley and Daroff's Neurology, Fig. 106.19)
Key steps from the flowchart:
- Patient fulfills clinical criteria → proceed to NCS + CSF
- If NCS/CSF normal → repeat in 1 week; consider other diagnoses
- If abnormal → confirmed diagnosis
- Monitor bulbar function, BP, FVC every 4-6 hours; assess severity
- Walks unassisted, no progression → Conservative management (observe)
- Walks with support OR bedridden AND symptoms <30 days → IVIG or plasma exchange
- FVC <12-15 mL/kg → Mechanical ventilation
7. IVIG vs Plasmapheresis - Comparative Details
| Feature | IVIG | Plasma Exchange (PE) |
|---|---|---|
| Dose | 0.4 g/kg/day × 5 days (total 2 g/kg) | 5 sessions over 10-14 days (200-250 mL/kg total) |
| Efficacy | Equal to PE | Equal to IVIG |
| Preferred? | Yes - easier, safer, more accessible | Alternative if IVIG unavailable/contraindicated |
| Mechanism | Suppresses immune response; Fc receptor blockade; anti-idiotype antibodies | Removes circulating antibodies, complement, cytokines |
| Contraindications | IgA deficiency (anaphylaxis risk); renal failure | Haemodynamic instability; sepsis; poor venous access |
| Side effects | Headache, fever, rash, renal failure (sucrose-containing), thrombosis, aseptic meningitis | Hypotension, line complications, coagulopathy |
| Combination with each other | NO benefit; not recommended | Same |
| Corticosteroids combined | No added benefit; may worsen | Same |
Timing matters: Treatment within 2 weeks of onset is most effective; treatment up to 4 weeks is still beneficial. After 4 weeks, immunotherapy is unlikely to help.
8. Pain Management in GBS (Often Undertreated)
Pain is present in ~70% of GBS patients and persists in 1/3 for up to a year:
| Pain Type | Best Treatment |
|---|---|
| Neuropathic (burning, dysesthetic) | Gabapentin, carbamazepine, pregabalin |
| Deep aching/musculoskeletal | NSAIDs, opioids |
| Severe acute pain | Opioids (morphine, tramadol) |
| Epidural pain | Epidural infusion (in ventilated patients) |
9. Rehabilitation in GBS (Post-Acute Phase)
A 2026 systematic review (PMID 41824781) confirms rehabilitation is essential in GBS recovery:
- Physiotherapy: progressive resistance and aerobic exercise; reduces fatigue and improves functional capacity
- Occupational therapy: activities of daily living retraining
- Speech therapy: bulbar and swallowing rehabilitation
- Respiratory physiotherapy: breathing exercises, weaning protocols
- Psychological support: depression and anxiety are common; quality of life is significantly affected
- Orthoses: ankle-foot orthoses for foot drop; wrist splints
10. GBS vs Acute-Onset CIDP - A Critical Distinction
Some patients initially diagnosed with GBS actually have acute-onset CIDP - this matters because CIDP responds to steroids but GBS does not.
Suspect acute-onset CIDP (not GBS) if:
- Disease progresses or deteriorates beyond 8 weeks
- >2 treatment-related fluctuations (improvement then worsening after IVIG/PE), especially beyond 1 month
- Prominent sensory symptoms dominate
- Multifocal enlargement of peripheral nerves on ultrasound
- >3 treatment-related fluctuations is almost diagnostic of CIDP
GBS recurrence: Up to 5% of GBS patients have a true recurrence after complete recovery - this is rare and distinct from treatment-related fluctuations.
11. Prognostic Scoring - EGOS / mEGOS
The Erasmus GBS Outcome Score (EGOS) predicts the probability of walking independently at 6 months:
| Predictor of Poor Outcome | Details |
|---|---|
| Age >60 years | Higher score = worse |
| Preceding diarrhoeal illness | C. jejuni link |
| MRC sum score at admission | Lower = worse |
| Rapid progression (<7 days to peak) | Suggests axonal |
| Low distal CMAP amplitudes (<20% LLN) or inexcitable nerves | Axonal loss |
| Preceding CMV infection | Typically more severe |
| Ventilatory support needed | Severe disease |
| Hyponatremia | Autonomic/SIADH |
Higher CSF neurofilament light chain (NfL) and tau levels at admission predict worse motor and functional outcomes - emerging biomarkers for severity stratification.
12. Long-Term Outcomes
| Outcome | Proportion |
|---|---|
| Complete or near-complete recovery | ~80% |
| Recovery complete by 12 months | ~70% |
| Recovery complete by 24 months | ~82% |
| Persistent disability at 1 year | ~20% |
| Mortality (modern ICU care) | 2-5% |
| Recurrence | ~5% |
Causes of death in GBS:
- Respiratory failure / ventilator complications
- Autonomic instability (cardiac arrhythmias, sudden cardiac death)
- Pulmonary embolism
- Sepsis / pneumonia (aspiration)
- Acute hypoxic-ischaemic encephalopathy
13. GBS in Special Populations
GBS in Children
- More common form: AMAN (particularly in Asia after C. jejuni)
- MRI spine may show gadolinium enhancement of cauda equina
- Generally better prognosis than adults
- Pain and back pain are prominent early features
GBS in Pregnancy
- Rare but serious
- Physiological changes increase risk of respiratory failure and aspiration
- IVIG is preferred over plasma exchange (haemodynamic safer)
- Does not cross placenta; fetus generally unaffected
- Succinylcholine is contraindicated (risk of hyperkalaemia and cardiac arrest due to upregulated ACh receptors)
GBS in the Elderly
- Higher incidence (3.2 per 100,000 in >60 years)
- More severe disease; greater respiratory failure risk
- Poor prognosis; higher mortality
- Autonomic complications more dangerous
Summary Table: GBS Subtypes at a Glance
| Feature | AIDP | AMAN | AMSAN | MFS |
|---|---|---|---|---|
| Frequency (West) | 97% | Rare | Rare | 5-6% |
| Frequency (Asia) | Less common | Most common | Less common | 18% (Taiwan) |
| Motor weakness | Yes | Yes | Yes | Usually spared |
| Sensory loss | Yes (mild) | No | Yes | Ataxia |
| Ophthalmoplegia | Rare | Rare | Rare | Yes |
| Reflexes | Absent | Absent | Absent | Absent |
| Motor NCS | Slowed, blocks | Normal CV, low CMAP | Normal CV, low CMAP | Normal |
| SNAP | Abnormal | Normal | Absent | Reduced |
| Key antibody | None specific | Anti-GM1/GD1a | Anti-GD1b | Anti-GQ1b |
| Pathology | Demyelination | Motor axon/node | Motor + sensory axon | Sensory axon |
| Prognosis | Good | Good (often) | Poor | Excellent |
- Bradley and Daroff's Neurology in Clinical Practice, p. 2663-2675
- Goldman-Cecil Medicine, p. 4065-4068
This is a shared conversation. Sign in to Orris to start your own chat.