Rabdomyosarcoma
Reading File
Finding Sources
Searching PubMed
rhabdomyosarcoma
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have all the material needed for a comprehensive answer. Here it is:
Rhabdomyosarcoma (RMS)
Definition and Epidemiology
Rhabdomyosarcoma is a malignant mesenchymal tumor with skeletal muscle differentiation - the most common soft-tissue sarcoma of childhood. It accounts for 3-4.5% of all pediatric malignancies. Importantly, pediatric RMS frequently arises in locations that do not normally contain skeletal muscle (sinuses, head/neck, genitourinary tract), underscoring that these tumors arise from undifferentiated mesenchymal stem cells, not from mature muscle.
- Peak incidence: before age 20 (embryonal/alveolar subtypes)
- Pleomorphic subtype: occurs in adults
- ~35% of RMS cases present in the head and neck region
Histologic Subtypes
Four subtypes are recognized (Robbins & Kumar Basic Pathology):
| Subtype | Frequency | Age | Key Features |
|---|---|---|---|
| Embryonal | ~50% | Children/adolescents | Soft gray mass; primitive round/spindle cells in myxoid stroma; rhabdomyoblasts with strap-like cytoplasm and cross-striations |
| Alveolar | ~20% | Children/adolescents | Fibrous septa divide cells into alveolar clusters; uniform round cells with little cytoplasm; PAX3/PAX7-FOXO1 fusion |
| Pleomorphic | ~20% | Adults | Large, bizarre multinucleate eosinophilic cells; occurs on extremities |
| Spindle cell/sclerosing | ~10% | All ages | -- |
Embryonal RMS - Histology
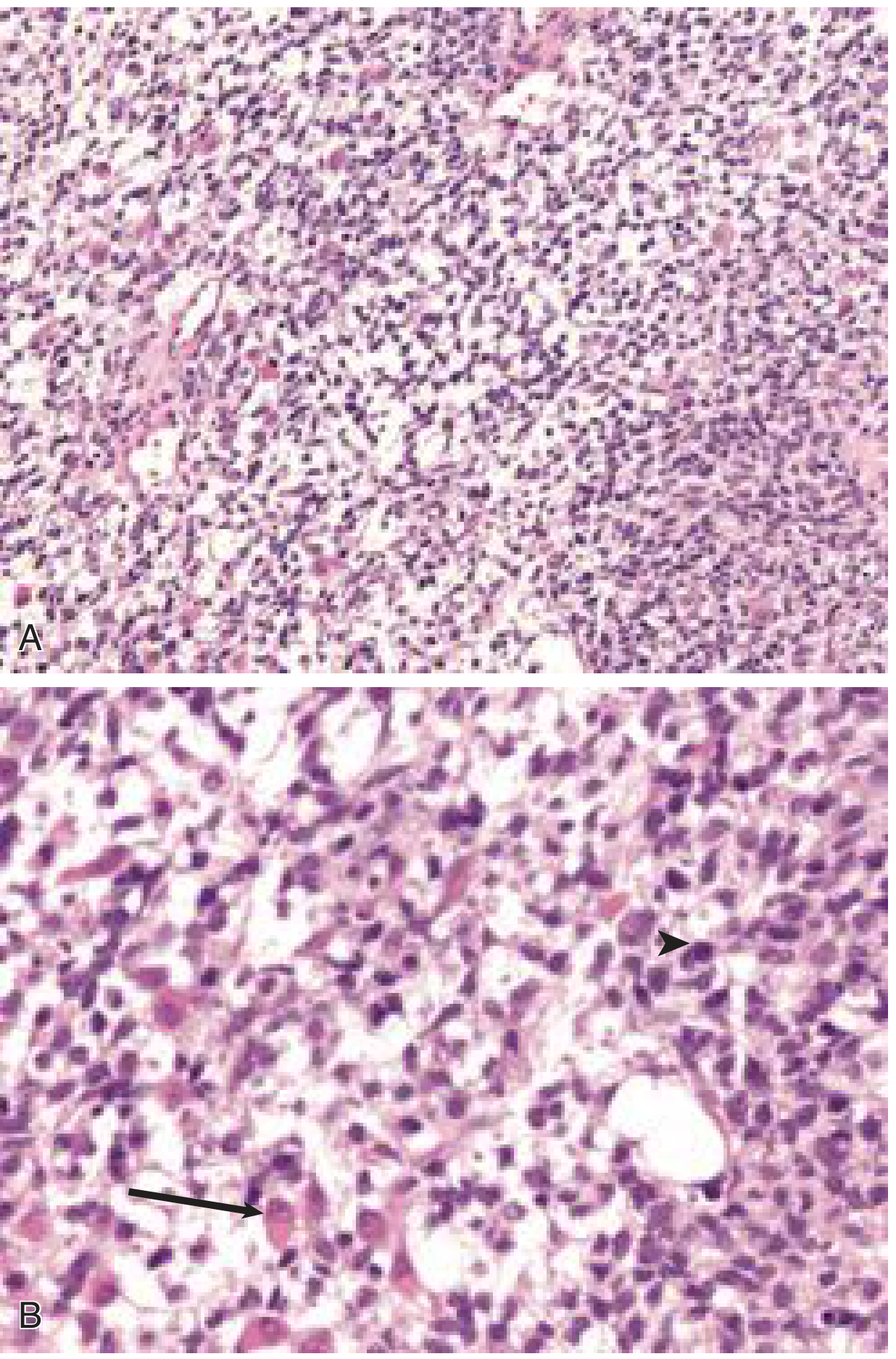
Fig. 1 - Embryonal RMS: (A) low power showing stellate and spindle cells in myxoid background; (B) high power showing a rhabdomyoblast with eccentric nucleus and eosinophilic cytoplasm (arrow) and a mitotic figure (arrowhead). - Cummings Otolaryngology
Sarcoma botryoides is a variant of embryonal RMS that develops as polypoid masses in the walls of hollow viscera (urinary bladder, vagina).
Alveolar RMS - Histology
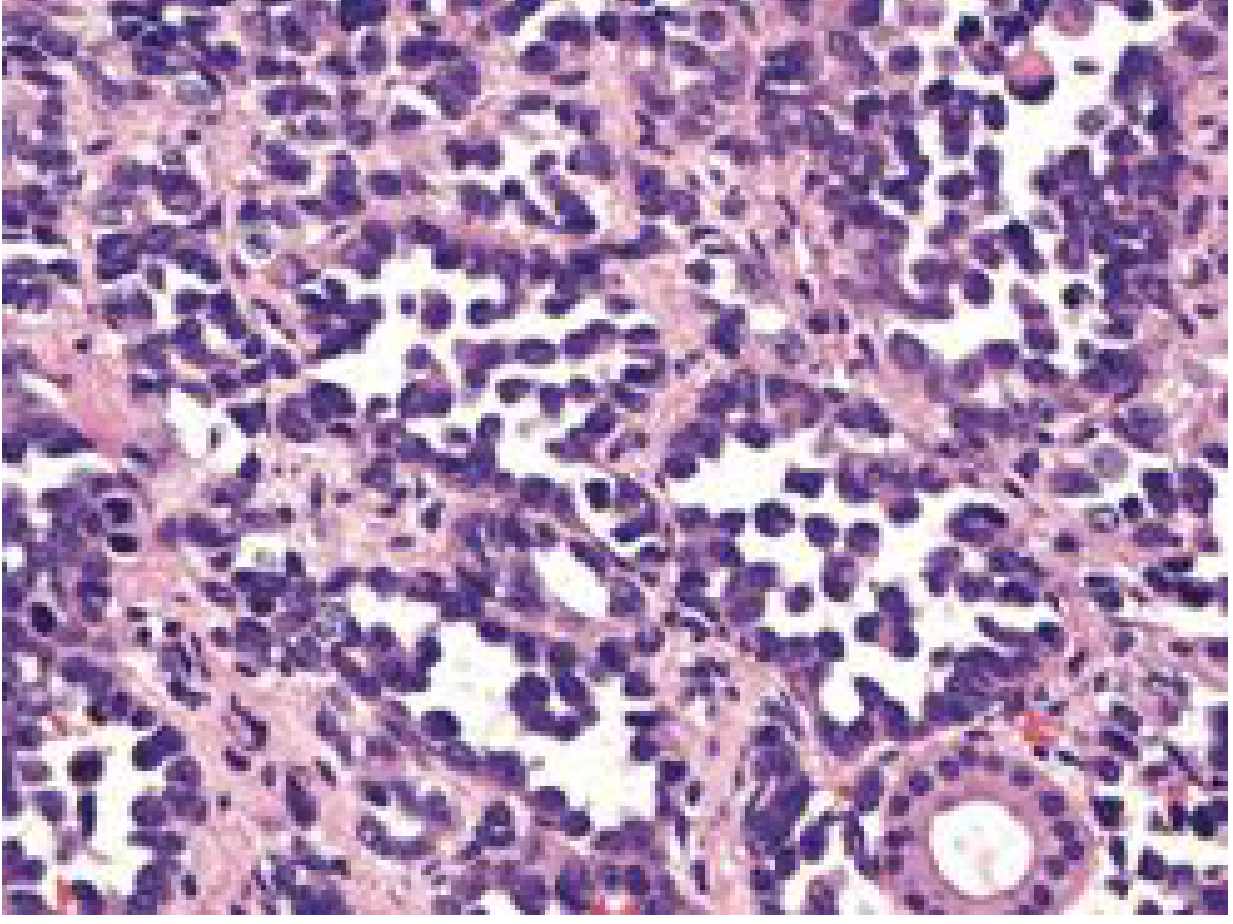
Fig. 2 - Alveolar RMS: small round blue cells in a dense configuration around alveolar-like spaces. - Cummings Otolaryngology
Embryonal vs. Alveolar (Robbins Pathology)
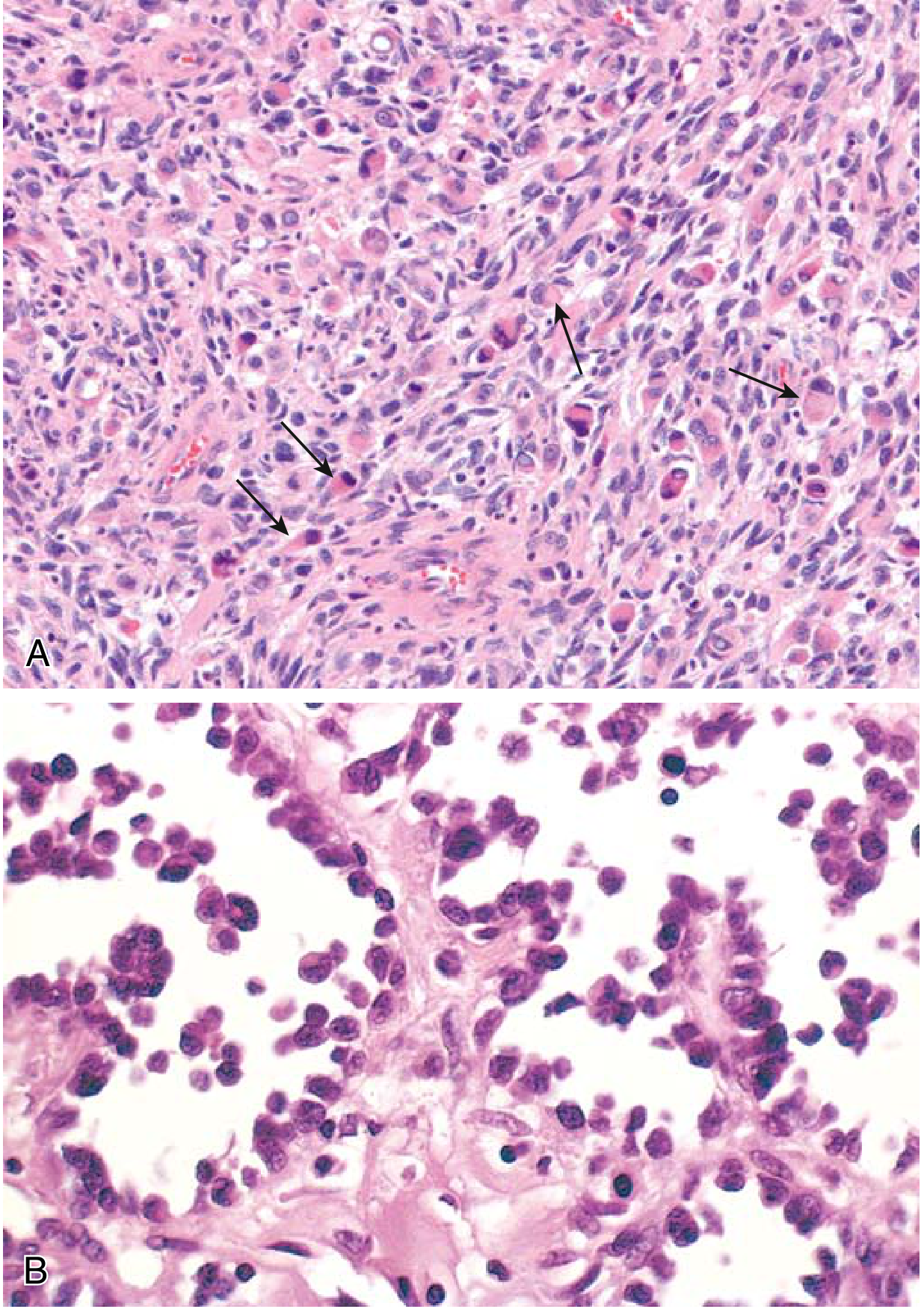
Fig. 3 - (A) Embryonal RMS: cells ranging from primitive/round to densely eosinophilic with skeletal muscle differentiation (arrows). (B) Alveolar RMS: numerous spaces lined by dyscohesive, uniform round tumor cells. - Robbins & Kumar Basic Pathology
Molecular Genetics
- Alveolar RMS carries characteristic chromosomal translocations:
- t(2;13) → PAX3-FOXO1 fusion (most common, worse prognosis)
- t(1;13) → PAX7-FOXO1 fusion (less common)
- PAX3 is a transcription factor that initiates skeletal muscle differentiation. The chimeric fusion protein interferes with normal differentiation (analogous to fusion proteins in acute leukemias) and drives aggressive behavior.
- Embryonal and pleomorphic subtypes are genetically heterogeneous (no defining translocation).
Clinical Presentation
RMS in the head and neck typically presents as a firm, asymptomatic mass. Symptoms reflect local mass effect and vary by site:
- Nasal obstruction, rhinorrhea, sinusitis, epistaxis
- Otitis media, aural polyps, otorrhea
- Hoarseness, epiphora
- Cranial nerve deficits if skull base erosion occurs
Common anatomical sites:
- Orbit (~25% of head/neck RMS) - favorable site
- Parameningeal sites (50% of head/neck RMS): middle ear/mastoid, nasal cavity, parapharyngeal space, paranasal sinuses, pterygopalatine/infratemporal fossa - higher risk of intracranial extension
- Genitourinary tract
- Extremities (pleomorphic type)
Metastatic spread:
- Regional lymph node involvement: ~12%
- Distant metastasis at diagnosis: 5-15% (primarily lungs and bone)
- Bone marrow involvement can occur
Diagnosis and Workup
Pathologic diagnosis:
- Small round blue cell tumor of childhood (differential includes Ewing sarcoma, lymphoma, neuroblastoma)
- IHC staining for muscle-specific proteins (myogenin, MyoD1, desmin) confirms skeletal myogenic lineage
- Rhabdomyoblasts with strap-like cytoplasm and visible cross-striations on light microscopy
- Cytogenetics/FISH for PAX-FOXO1 fusion (alveolar subtype)
Imaging:
- MRI - isointense to skeletal muscle on T1, high signal on T2; best for soft tissue extent and perineural spread
- CT - superior for bony erosion assessment near skull base
- FDG-PET/CT - sensitive/specific for macroscopic metastatic disease
- Technetium-99 bone scan - for osseous metastases
Laboratory and other workup:
- CBC, electrolytes, renal/liver function, coagulation studies
- Bone marrow aspirate (staging)
- Lumbar puncture for CSF cytology (parameningeal disease)
Staging
A two-system approach is used in cooperative group trials:
Pretreatment TNM Staging (summarized for Head/Neck RMS):
| Stage | Primary Site | Tumor | Size | Nodes | Metastasis |
|---|---|---|---|---|---|
| 1 | Favorable site (orbit, non-parameningeal) | T1 or T2 | Any | Any N | M0 |
| 2 | Unfavorable site | T1 or T2 | ≤5 cm | N0/NX | M0 |
| 3 | Unfavorable site | T1 or T2 | A: ≤5 cm; B: >5 cm | N1/Any | M0 |
| 4 | Any site | T1 or T2 | Any | Any | M1 |
Staging also incorporates:
- Surgical/clinical grouping (post-treatment extent of resection)
- Histologic subtype (alveolar = worse prognosis)
- Together these define the patient's prognostic risk stratification (low, intermediate, high risk) which drives treatment intensity.
Treatment
Management is multimodal - surgery, radiation, and chemotherapy:
-
Surgery: Wide local excision with clear margins when possible. For head/neck/orbital sites, function-preserving surgery is preferred. Mutilating surgery generally avoided.
-
Radiation: Indicated for most cases. Local control at the primary site, especially when margins are positive or for parameningeal disease.
- Recent evidence: A 2025 systematic review (PMID 40494941) compared photon radiotherapy vs. proton beam therapy in pediatric RMS - proton therapy may reduce toxicity while maintaining control.
-
Chemotherapy: Backbone regimen is VAC (Vincristine, Actinomycin D, Cyclophosphamide). Treatment stratified by risk group under Children's Oncology Group (COG) protocols.
-
Enrollment in clinical trials is strongly encouraged for all pediatric patients.
Prognosis
- 1960s: 5-year OS ~50%
- By 1990s (with multimodal chemo): ~70%
- Current outcomes highly dependent on risk stratification:
- Low risk (favorable histology, localized, complete resection): >90% 5-year OS
- Intermediate risk: ~60-80%
- High risk (metastatic or alveolar with poor features): <30%
- Alveolar RMS (especially PAX3-FOXO1 fusion) carries a worse prognosis than embryonal RMS
Key Associations
| Feature | Detail |
|---|---|
| Most common pediatric soft tissue sarcoma | Yes |
| Most common H&N subsite | Orbit |
| Worst prognosis subtype | Alveolar (PAX3-FOXO1) |
| Best prognosis subtype | Embryonal (especially botryoid variant) |
| Cooperative group | Children's Oncology Group (COG), former IRSG |
| IHC markers | Myogenin, MyoD1, Desmin, Muscle-specific actin |
Sources: Robbins & Kumar Basic Pathology; Cummings Otolaryngology Head and Neck Surgery; Campbell's Operative Orthopaedics 15th Ed.; Grainger & Allison's Diagnostic Radiology
Recent literature note: A 2025 meta-analysis (PMID 40494941) found comparable oncologic control with proton beam therapy vs. photon radiotherapy in pediatric RMS, with a potential toxicity benefit for protons - relevant for treatment planning at specialized centers.
Rhabdomyosarcoma eye
Finding Sources
Searching PubMed
orbital rhabdomyosarcoma
Finding Sources
Searching PubMed
orbital rhabdomyosarcoma children treatment
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive textbook content and beautiful clinical images. Here is the complete, detailed answer:
Orbital Rhabdomyosarcoma (RMS)
Overview
Orbital RMS is the most common primary orbital malignancy in children and the most common soft-tissue sarcoma of childhood affecting the orbit. Despite this, it remains a rare condition overall. Key facts:
- ~90% occur in children under 16 years; average age of onset is 7 years
- The orbit accounts for ~25% of all head/neck RMS cases, and is classified as a favorable site due to its excellent response to treatment
- The tumor arises from undifferentiated mesenchymal cells with the potential to differentiate into striated muscle - NOT from mature extraocular muscles
Histologic Subtypes in the Orbit
| Subtype | Orbital Frequency | Prognosis |
|---|---|---|
| Embryonal | 85% of orbital lesions | Good |
| Alveolar | Most of the remaining ~15% | Worse; PAX3/PAX7-FOXO1 fusions |
| Botryoid | ~4% | Good (botryoid = best prognosis) |
| Pleomorphic | Rare in orbit | -- |
Histology - Fig. 4.42A (Kanski's Clinical Ophthalmology)
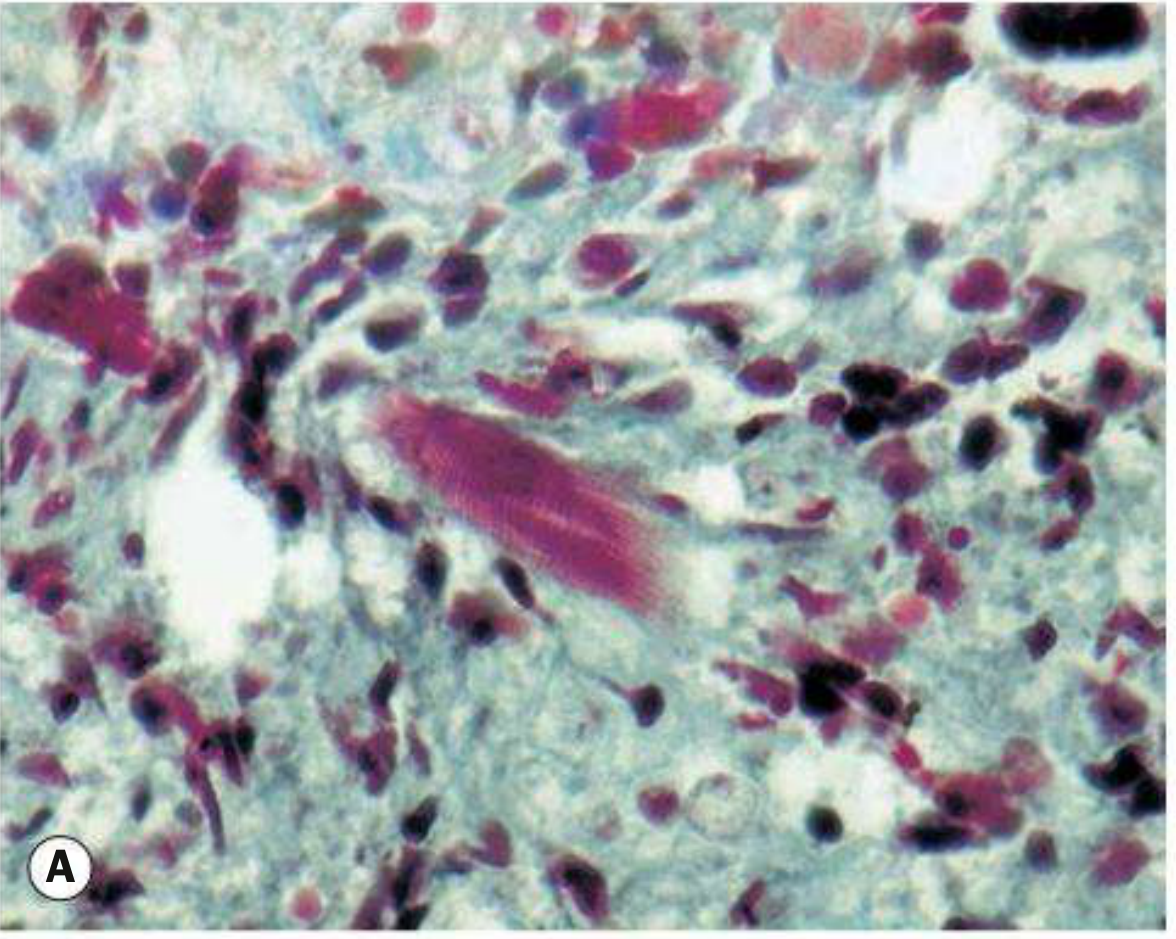
Fig. 4.42A - Orbital RMS histology (Masson trichrome stain). The differentiated rhabdomyoblast in the center of the field shows characteristic cross-striations confirming skeletal muscle lineage. - Kanski's Clinical Ophthalmology 10th Ed.
Clinical Presentation
Symptoms
- Rapidly progressive unilateral proptosis - the hallmark presentation
- Can mimic orbital cellulitis (KEY: RMS can mimic orbital infection in a child)
- Diplopia is frequent; pain is less common
- Swelling and redness of overlying skin, but skin is not warm (helps distinguish from true cellulitis)
Signs - Location
- Most commonly superonasal or superior in position (Fig. B below)
- Can arise anywhere in the orbit, including inferiorly (Fig. C)
- Rarely arises in conjunctiva or uvea
Clinical Photos - Fig. 4.42B (Kanski's)
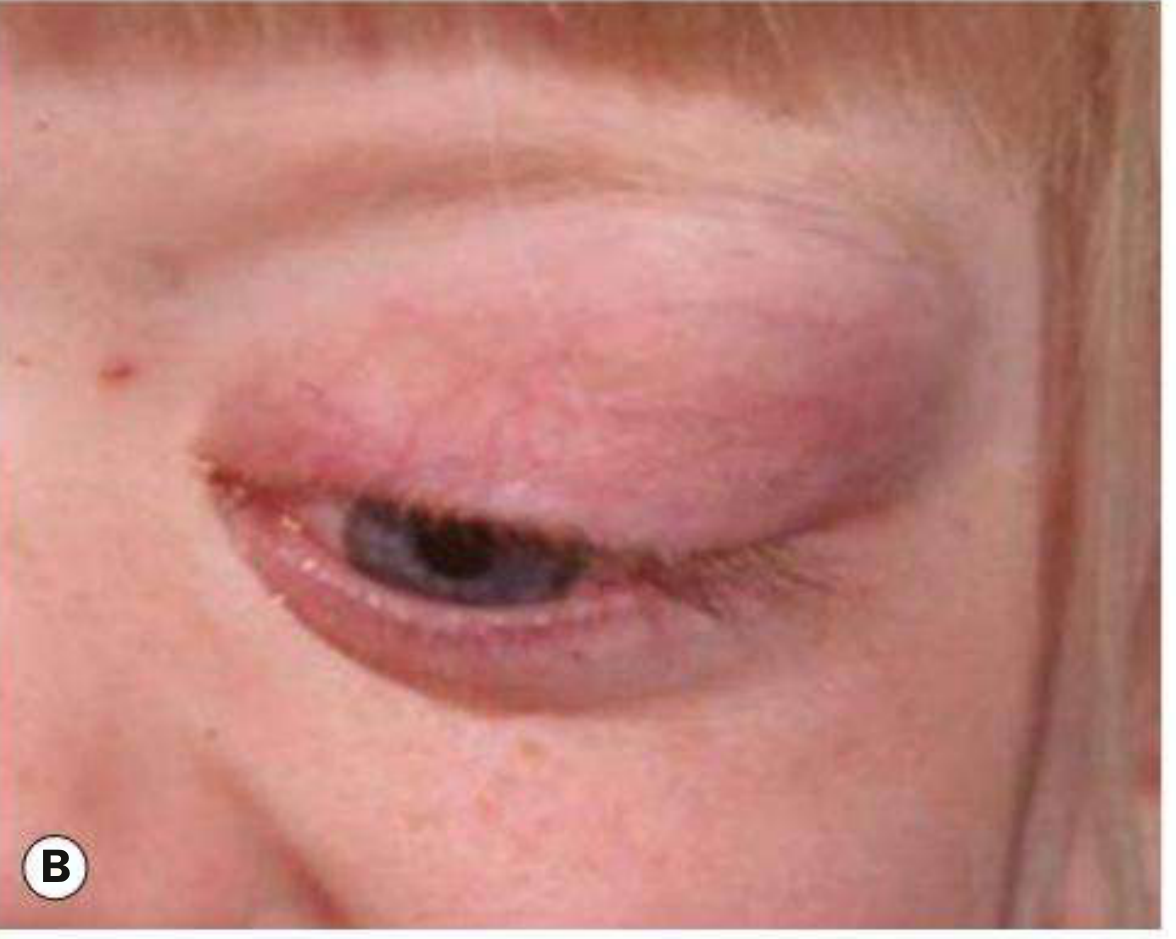
Fig. 4.42B - Orbital RMS: massive superior orbital lesion causing proptosis with marked lid swelling and erythema. Note the inflammatory appearance that can easily be mistaken for orbital cellulitis. - Kanski's Clinical Ophthalmology 10th Ed.
Differential Diagnosis of Acute Proptosis in a Child
When RMS presents rapidly, the differential includes:
- Orbital cellulitis / abscess (most important mimic)
- Lymphangioma with acute hemorrhage
- Leukemia / granulocytic sarcoma
- Metastatic neuroblastoma
- Langerhans cell histiocytosis
- Lymphoproliferative disease
- Dermoid cyst (leaking)
Clinical pearl (Wills Eye Manual): In cases of acute onset + rapid progression + mass on imaging in a child, emergency incisional biopsy is indicated to rule out aggressive malignancy such as RMS.
Imaging
CT Scan
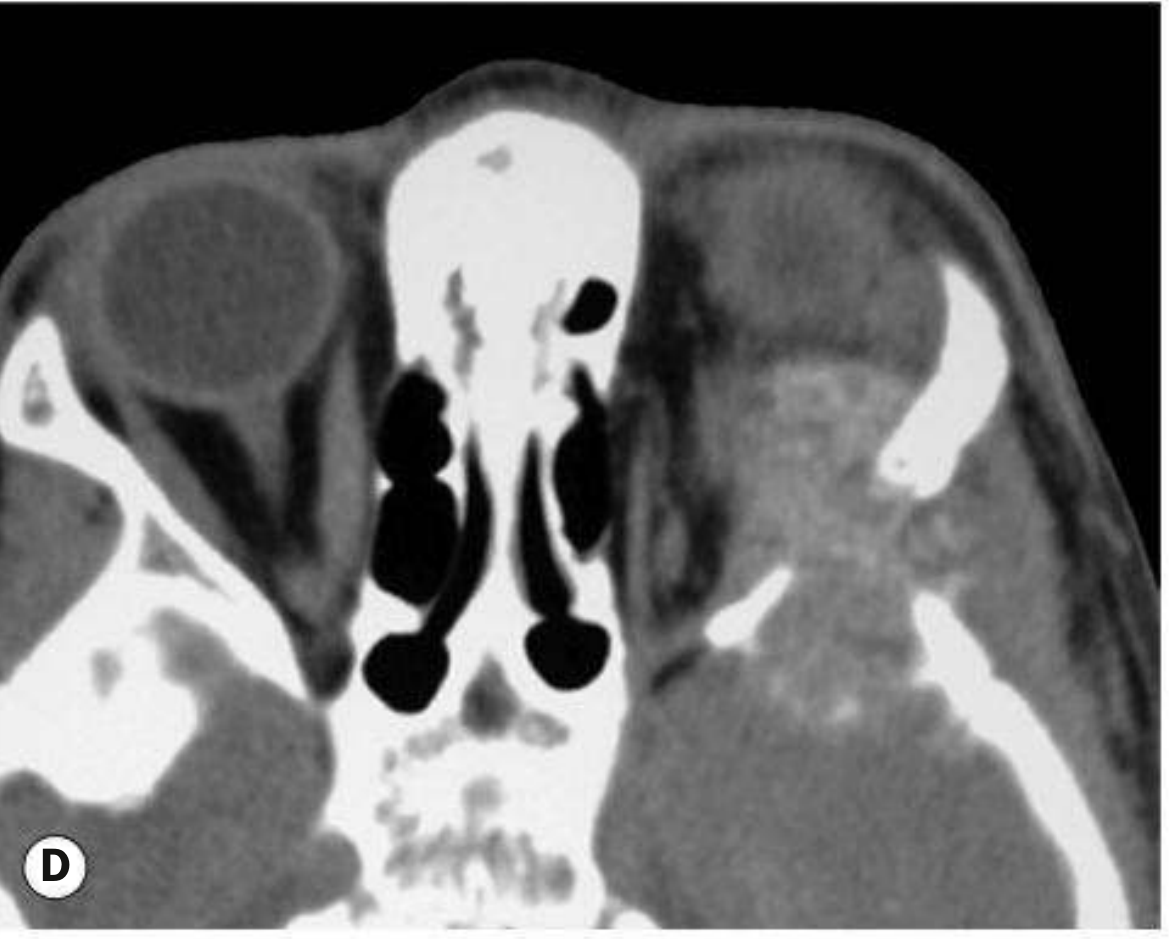
Fig. 4.42D - Axial CT showing a large orbital RMS with bony destruction and intracranial extension. - Kanski's Clinical Ophthalmology 10th Ed.
| Modality | Findings |
|---|---|
| CT | Poorly defined mass of homogeneous density; often with adjacent bony destruction; best for bony erosion assessment |
| MRI | Poorly defined mass; isointense to muscle on T1, hyperintense to muscle on T2; better for soft-tissue extent, perineural spread |
Workup and Staging
Biopsy is required - incisional biopsy provides:
- Histopathological subtype confirmation
- Cytogenetic characteristics (PAX-FOXO1 fusion status)
- Frozen section for urgent cases
Systemic staging workup includes:
- Physical exam (lymph node palpation)
- Chest and bone radiographs
- Bone marrow aspiration
- Lumbar puncture
- Liver function studies
- FDG-PET/CT (sensitive for macroscopic metastases)
- Technetium-99 bone scan (osseous metastases)
Most common metastatic sites: Lung and bone
Staging system: Intergroup Rhabdomyosarcoma Study Group (IRSG) TNM + surgical group system, now under Children's Oncology Group (COG). The orbit is a Stage 1 favorable site (any T, any N, M0).
Treatment
Treatment is multimodal - never primary exenteration:
| Step | Details |
|---|---|
| 1. Urgent biopsy | Confirm diagnosis, subtype, cytogenetics |
| 2. Systemic chemotherapy | VAC regimen (Vincristine, Actinomycin D, Cyclophosphamide) - used for all cases and for disseminated disease |
| 3. Local radiotherapy | For localized orbital lesions; used for local control after chemo induction |
| 4. Surgical debulking | Only occasionally for well-defined orbital lesion; not routine |
Key evolution: Orbital exenteration (removal of the eye and orbit contents) was once the standard of care - it is no longer indicated. Multimodal chemo + radiation has replaced it with equivalent or superior outcomes while preserving vision and the globe.
Emerging therapies:
- Entinostat (HDAC inhibitor) with chemotherapy may improve prognosis in selected cases, but is not yet approved
- The oncogenic PAX3-FOXO1 fusion gene is associated with treatment resistance and a 40-45% decrease in overall survival - its presence must be identified at diagnosis
- Immunotherapy (e.g., rituximab) is under investigation
Prognosis
- Orbital lesions have the BEST prognosis among all RMS sites
- Localized orbital disease: ~90% long-term survival
- Prognosis decreases substantially with spread to adjacent anatomy:
- Paranasal sinus extension - worse
- Intracranial extension - significantly worse
- Embryonal subtype: better prognosis than alveolar
- Overall long-term prognosis has greatly improved over the past 50 years due to advances in chemo and radiotherapy
Late effects to monitor:
- Dental and bone alterations after orbital RMS treatment (radiation-related)
- Orbital growth abnormalities (radiation-induced)
- Cataract, radiation retinopathy
Comparison: Adult vs. Pediatric Orbital RMS
A 2025 comparative study (PMID 39792629) compared adult and pediatric orbital RMS, highlighting that adult cases have different characteristics and outcomes than the pediatric form - adults tend to present with less favorable histology and worse outcomes.
A 2025 long-term outcomes study (PMID 40002210) from a multidisciplinary center confirmed good long-term outcomes with modern combined-modality treatment for pediatric orbital RMS.
Sources: Kanski's Clinical Ophthalmology, A Systematic Approach 10th Ed.; Wills Eye Manual - Office and Emergency Room Diagnosis and Treatment; Cummings Otolaryngology Head and Neck Surgery; Goldman-Cecil Medicine
Shirmers test
Finding Sources
Searching PubMed
Schirmer test dry eye diagnosis
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Excellent - I have everything I need. Here is a comprehensive, image-rich answer:
Schirmer Test
What It Is
The Schirmer test is a clinical assessment of aqueous tear production (lacrimal secretion). It uses a standardized strip of filter paper placed in the lower conjunctival fornix to measure the amount of wetting over 5 minutes.
Equipment
- No. 41 Whatman filter paper strip - 5 mm wide × 35 mm long
- Folded 5 mm from one end to create a hook that hangs over the lid margin
Clinical Photo
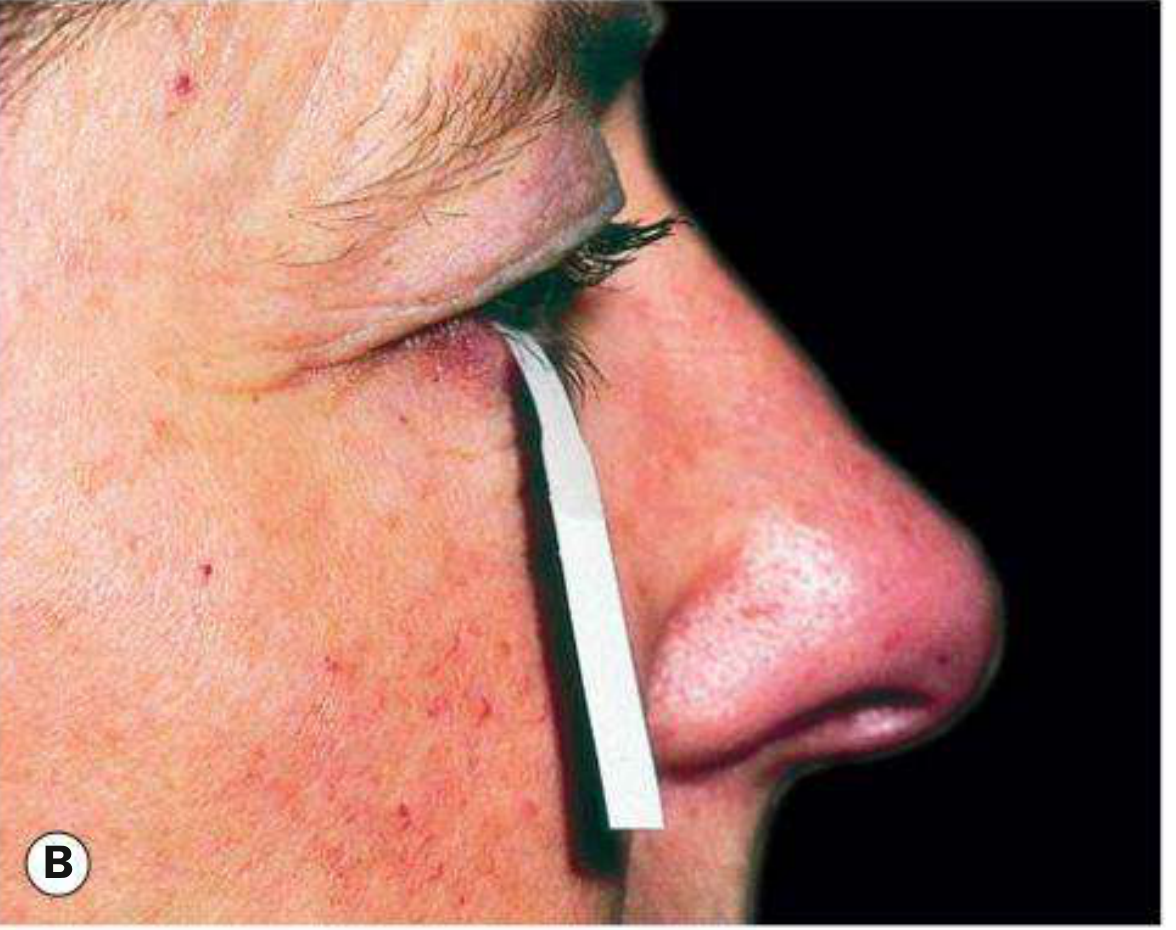
Fig. 5.8B - Schirmer test: filter paper strip inserted at the junction of the middle and outer third of the lower lid, with the patient's eyes gently closed. - Kanski's Clinical Ophthalmology 10th Ed.
Two Variants
| Schirmer 1 | Schirmer 2 | |
|---|---|---|
| Anesthesia | Without topical anesthesia | With topical anesthesia |
| What it measures (in theory) | Maximum basic + reflex secretion | Basic secretion only |
| Abnormal result | < 10 mm wetting in 5 minutes | < 6 mm wetting in 5 minutes |
| Practical note | Tear reflex stimulated by paper itself | Topical anesthesia cannot abolish all sensory/psychological stimuli - so Schirmer 2 is not a pure basic secretion test in practice |
Note from Cummings Otolaryngology: In the pre-surgical ptosis setting (assessing dry eye before ptosis correction), < 5 mm wetting with topical anesthetic raises concern for significant dry eye and potential post-operative complications.
Technique (Step by Step)
- Dry the conjunctival sac - delicately blot excess tears from the inferior fornix
- If using topical anesthesia (Schirmer 2), also remove excess anesthetic with filter paper
- Fold the strip 5 mm from one end
- Insert the folded end at the junction of the middle and outer third of the lower lid - take care not to touch the cornea or lashes
- Ask the patient to keep eyes gently closed
- Remove after 5 minutes and measure the length of wetting from the fold
Interpretation
| Wetting (5 min) | Without anesthesia | With anesthesia |
|---|---|---|
| Normal | ≥ 10 mm | ≥ 6 mm |
| Abnormal | < 10 mm | < 6 mm |
| Severe dry eye concern (pre-op) | -- | < 5 mm |
Important limitations:
- Results are variable - a single Schirmer test should NOT be the sole criterion for diagnosing dry eye
- Repeatedly abnormal results are highly supportive of the diagnosis
- Cannot perfectly separate basic from reflex secretion even with anesthesia
Clinical Indications
- Diagnosis of dry eye disease (keratoconjunctivitis sicca, KCS)
- Sjögren syndrome workup - Schirmer test combined with Rose Bengal staining; the Rose Bengal stain is reported to be more specific for KCS in this context
- Pre-operative assessment before ptosis surgery, blepharoplasty, or other periocular procedures
- Monitoring response to dry eye treatment
Dry Eye Diagnostic Battery (Schirmer in Context)
The Schirmer test is one of several tests used together to evaluate dry eye:
| Test | What It Measures |
|---|---|
| Schirmer test | Aqueous tear volume / secretion |
| Tear film break-up time (TBUT) | Tear film stability (< 10 sec = abnormal) |
| Rose Bengal / Lissamine green staining | Dead/devitalized epithelial cells; mucin layer integrity |
| Fluorescein staining | Corneal/conjunctival epithelial damage |
| Tear osmolarity (TearLab) | Hyperosmolarity confirms dry eye (> 308 mOsm/L) |
- Interpalpebral staining pattern on Rose Bengal/Lissamine green suggests aqueous tear deficiency
- Fluorescein stains areas where damage has allowed dye entry into tissues
Associated Conditions
Abnormal Schirmer test is found in:
- Sjögren syndrome (primary and secondary)
- Age-related lacrimal gland atrophy
- Lacrimal gland destruction (sarcoidosis, lymphoma, prior radiation)
- Cicatricial conjunctivitis (trachoma, pemphigoid, Stevens-Johnson)
- Riley-Day syndrome (familial dysautonomia)
- Post-ptosis surgery / blepharoplasty complications
- Contact lens wear
Sources: Kanski's Clinical Ophthalmology, A Systematic Approach 10th Ed.; Cummings Otolaryngology Head and Neck Surgery
This is a shared conversation. Sign in to Orris to start your own chat.