biochemical basis of parkinson and alzeimer disease
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Parkinson disease" AND "alpha-synuclein" AND pathogenesis
Searching PubMed
"Alzheimer disease" AND amyloid AND tau AND pathogenesis
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Now I have comprehensive textbook content and images. Let me compile the full, detailed answer.
Biochemical Basis of Parkinson's Disease and Alzheimer's Disease
PARKINSON'S DISEASE (PD)
Core Neuropathology
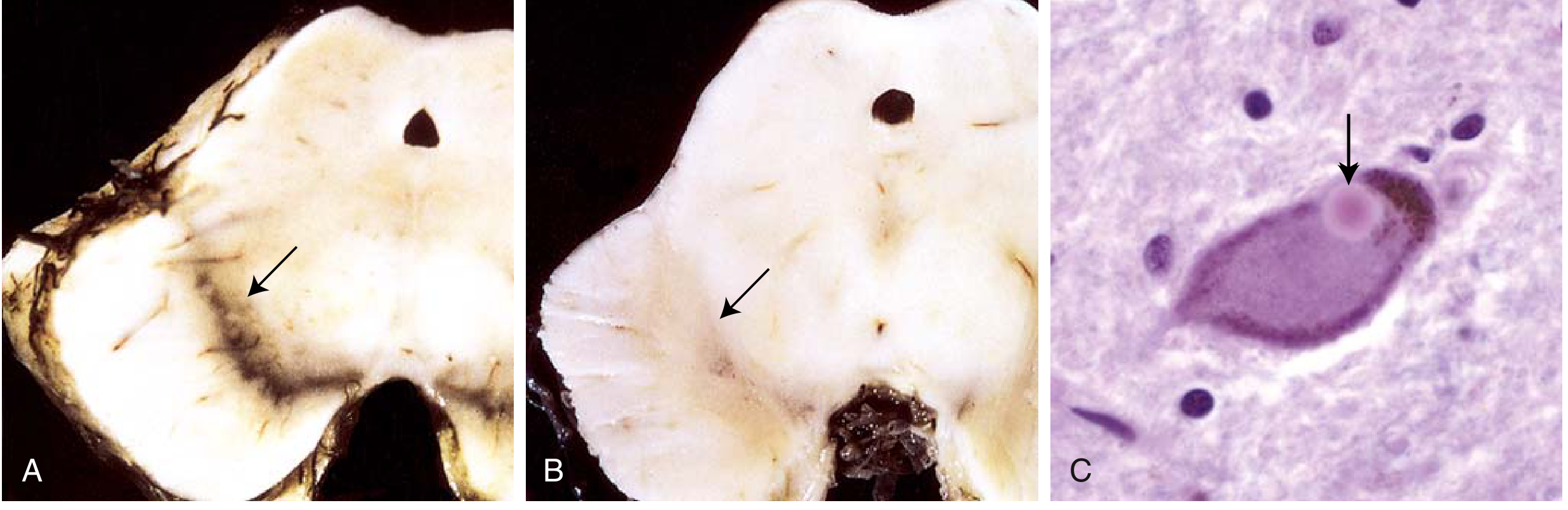
PD is a neurodegenerative disorder in which the most prominent pathology involves the pigmented dopaminergic neurons of the substantia nigra (pars compacta). These neurons project to the striatum (nigrostriatal pathway) and are essential for motor control. Their progressive loss leads to the classic motor triad of tremor, rigidity, and bradykinesia.
At autopsy, the substantia nigra is visibly depigmented (loss of neuromelanin). Microscopically, there is neuronal dropout with reactive gliosis.
Key Biochemical Mechanisms
1. Alpha-Synuclein Aggregation and Lewy Bodies
The hallmark lesion of PD is the Lewy body - a round, eosinophilic, cytoplasmic inclusion found in surviving neurons. Lewy bodies consist of:
- Alpha-synuclein (alpha-syn) - a normally soluble synaptic protein involved in vesicle trafficking and synaptic transmission
- Neurofilaments, ubiquitin, and other proteins
Normally, alpha-syn exists as a monomer and helps regulate dopamine release at presynaptic terminals. In PD, it misfolds and polymerizes into insoluble fibrillar aggregates (Lewy bodies and Lewy neurites). This is central to pathogenesis.
The spread of alpha-syn pathology follows a predictable anatomical pattern (Braak staging): it begins in the enteric nervous system and lower brainstem (olfactory bulb, dorsal motor nucleus of vagus), then ascends through the brainstem to the substantia nigra, and eventually reaches the cortex - explaining why non-motor symptoms (sleep disorders, constipation, anosmia) often precede motor features.
2. Impaired Protein and Organelle Clearance
Multiple lines of evidence point to failure of autophagy and lysosomal degradation as central mechanisms:
- Alpha-syn aggregates are normally cleared by autophagy
- Parkin - an E3 ubiquitin ligase - mutations cause autosomal recessive PD. Parkin tags damaged mitochondria for degradation (mitophagy via the PINK1/Parkin pathway)
- PINK1 (PTEN-induced kinase 1) mutations also cause recessive PD; PINK1 accumulates on depolarized mitochondria and recruits Parkin
- LRRK2 (leucine-rich repeat kinase 2) gain-of-function mutations are the most common cause of autosomal dominant PD. LRRK2 plays roles in endosomal trafficking and autophagy pathways
- Heterozygous mutations in glucocerebrosidase (GBA - the Gaucher disease gene, a lysosomal enzyme) are a major risk factor for sporadic PD, again implicating impaired lysosomal function
3. Mitochondrial Dysfunction and Oxidative Stress
- Complex I of the mitochondrial electron transport chain is reduced in the substantia nigra of PD patients
- MPTP (a toxin found in "synthetic" heroin) selectively destroys dopaminergic neurons by inhibiting Complex I, producing a PD-like syndrome - confirming the link between mitochondrial dysfunction and PD
- Dopamine metabolism itself generates reactive oxygen species (ROS), making dopaminergic neurons especially vulnerable to oxidative stress
- The accumulation of neuromelanin (the dark pigment of SN neurons) is a byproduct of dopamine oxidation; this may amplify local oxidative damage
4. Dopamine Deficiency - The Functional Consequence
Loss of nigral dopaminergic neurons depletes dopamine in the striatum (caudate/putamen). This disrupts the balance of the basal ganglia circuitry:
- The direct pathway (D1 receptors, excitatory) is underactive
- The indirect pathway (D2 receptors, inhibitory) becomes overactive
- Net result: increased inhibitory output from the basal ganglia to the thalamus, reducing thalamocortical activation and manifesting as the akinesia/bradykinesia of PD
This explains why L-DOPA (the dopamine precursor) remains the primary treatment - it replenishes striatal dopamine.
5. Genetic Mutations Summary
| Gene | Inheritance | Function |
|---|---|---|
| SNCA (alpha-synuclein) | Autosomal dominant | Synaptic protein; point mutations or duplications cause PD |
| LRRK2 | Autosomal dominant | Kinase; most common familial PD mutation |
| Parkin (PARK2) | Autosomal recessive | E3 ubiquitin ligase; mitophagy |
| PINK1 | Autosomal recessive | Mitochondrial kinase; mitophagy |
| GBA | Risk factor | Lysosomal glucocerebrosidase |
ALZHEIMER'S DISEASE (AD)
Core Neuropathology
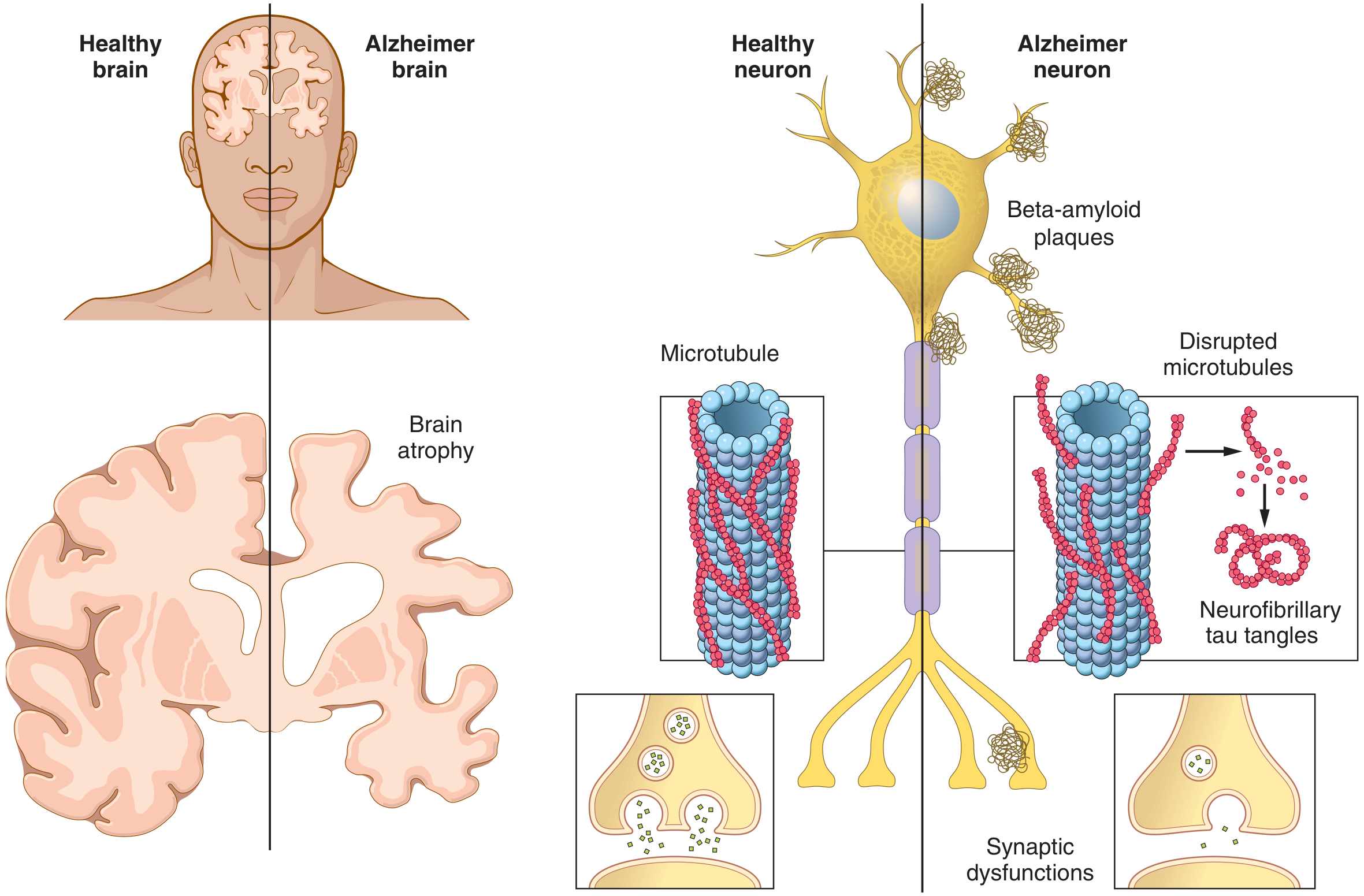
AD is the most common neurodegenerative cause of dementia. The two defining pathological hallmarks are:
- Extracellular amyloid-beta (Abeta) plaques - neuritic/senile plaques
- Intracellular neurofibrillary tangles (NFTs) - composed of hyperphosphorylated tau protein
These accumulate predominantly in the hippocampus, entorhinal cortex, association cortices, and basal forebrain, leading to progressive memory loss, language impairment, and visuospatial deficits.
Key Biochemical Mechanisms
1. Amyloid Precursor Protein (APP) and Abeta Production
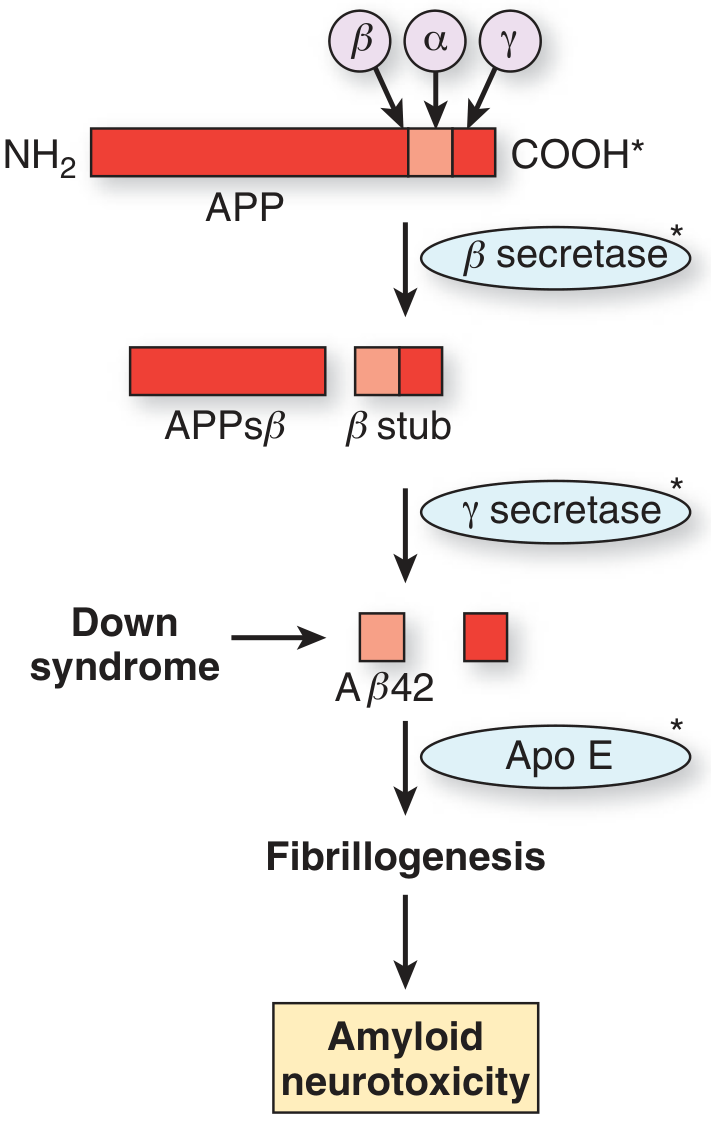
The amyloid-beta peptide is a cleavage product of the Amyloid Precursor Protein (APP), a transmembrane protein normally bound to neuronal membranes. APP is cleaved by secretase enzymes:
- Non-amyloidogenic pathway: alpha-secretase cleaves within the Abeta sequence - produces soluble sAPP-alpha, which is neuroprotective. No Abeta is produced.
- Amyloidogenic pathway: sequential cleavage first by beta-secretase (BACE1), then by gamma-secretase (presenilin complex) generates:
- Abeta40 - more abundant, less aggregating
- Abeta42 - more hydrophobic and aggregation-prone; the main species in plaques
Abeta42 monomers aggregate into soluble oligomers (likely the most neurotoxic species), then protofibrils, and finally insoluble amyloid fibrils deposited as plaques. The soluble oligomers disrupt synaptic function, activate neuroinflammation, and trigger downstream tau pathology.
2. The Amyloid Cascade Hypothesis
Evidence supporting the central role of Abeta:
- All known familial AD mutations (APP, Presenilin-1, Presenilin-2) increase Abeta42 production or the Abeta42/40 ratio
- Presenilin-1 and 2 are the catalytic components of gamma-secretase - their mutations increase Abeta42 generation
- Down syndrome (trisomy 21) carries three copies of chromosome 21 where APP resides - virtually all Down syndrome patients develop AD pathology by midlife
- ApoE4 (apolipoprotein E allele epsilon-4) - the strongest genetic risk factor for sporadic AD - impairs Abeta42 clearance and promotes fibrillogenesis
- Anti-amyloid antibodies (e.g., lecanemab, donanemab) modestly slow cognitive decline in early AD, providing clinical validation of the target
However, the relationship between plaque load and neuronal loss is imperfect, and soluble oligomers rather than insoluble plaques may be the primary toxic agents.
3. Tau Hyperphosphorylation and Neurofibrillary Tangles
Tau is a microtubule-associated protein that normally promotes microtubule assembly and stabilizes the axonal cytoskeleton. In AD:
- Tau becomes hyperphosphorylated due to an imbalance between kinases (GSK-3beta, CDK5) and phosphatases
- Hyperphosphorylated tau dissociates from microtubules, causing them to disassemble - impairing axonal transport
- Free tau molecules aggregate into paired helical filaments (PHFs) that form neurofibrillary tangles (NFTs)
- NFTs follow a predictable Braak staging (I-VI), spreading from entorhinal cortex and hippocampus to association cortices - this spread correlates far more closely with cognitive decline than plaque burden
Tau pathology also triggers neuroinflammation, worsening neuronal injury. The close correlation of tau PET and temporal cortical atrophy with cognitive ability confirms tau as a critical driver of clinical disease.
4. Cholinergic Deficit - The Functional Consequence
A key functional deficit in AD is the loss of cholinergic neurons in the nucleus basalis of Meynert (basal forebrain), which provides cholinergic innervation to the hippocampus and cortex. This reduces:
- Choline acetyltransferase (ChAT) activity
- Acetylcholine (ACh) levels in the hippocampus and neocortex
The degree of cholinergic loss correlates with the degree of dementia. This is the basis for treating AD with acetylcholinesterase inhibitors (donepezil, rivastigmine, galantamine), which boost ACh levels in the synapse. In early AD, cholinergic postsynaptic targets remain intact, so this approach can still be beneficial.
5. Neuroinflammation
Amyloid plaques and NFTs activate microglia and astrocytes, generating an inflammatory cascade (cytokines, complement, reactive oxygen/nitrogen species) that accelerates neuronal death. Recent genome-wide association studies have identified risk genes (TREM2, CLU, CR1) that are expressed in microglia, further linking immune dysregulation to AD pathogenesis.
6. Genetic Factors Summary
| Gene/Factor | Role |
|---|---|
| APP (chr 21) | Abeta precursor; mutations increase Abeta42 |
| Presenilin-1 (PSEN1) | gamma-secretase component; most common early-onset familial AD mutation |
| Presenilin-2 (PSEN2) | gamma-secretase component; rarer autosomal dominant AD |
| APOE4 allele | Impairs Abeta clearance; greatest risk for sporadic AD |
| TREM2 | Microglial receptor; affects neuroinflammatory response |
COMPARISON: PD vs. AD
| Feature | Parkinson's Disease | Alzheimer's Disease |
|---|---|---|
| Primary site of pathology | Substantia nigra | Hippocampus, entorhinal cortex, cortex |
| Key protein aggregate | Alpha-synuclein (Lewy bodies) | Abeta plaques + tau NFTs |
| Neurotransmitter deficit | Dopamine | Acetylcholine (and others) |
| Main symptoms | Motor: tremor, rigidity, bradykinesia | Cognitive: memory loss, aphasia, apraxia |
| Key genetic mechanisms | Alpha-syn, LRRK2, Parkin/PINK1 (mitophagy) | APP, Presenilin, ApoE4 |
| Protein clearance failure | Autophagy/lysosomal, ubiquitin-proteasome | Secretase dysregulation, ApoE4-mediated |
| Mitochondrial involvement | Central (Complex I inhibition, PINK1/Parkin) | Secondary (Abeta disrupts mitochondria) |
| Overlap | ~25% of AD patients have Lewy body co-pathology | Lewy body dementia bridges PD and AD |
Shared Themes
Both diseases share several pathogenic principles:
- Misfolded protein aggregation - alpha-syn vs. Abeta/tau
- Failure of protein degradation - autophagy, lysosomal, and proteasomal dysfunction
- Mitochondrial dysfunction and oxidative stress
- Neuroinflammation - microglial and astrocyte activation
- Synaptic dysfunction preceding neuronal death
- Prion-like spread - both alpha-syn and tau appear to propagate in a template-directed manner through neuronal networks
Sources: Robbins & Kumar Basic Pathology; Guyton and Hall Textbook of Medical Physiology; Adams and Victor's Principles of Neurology, 12th Edition; Bradley and Daroff's Neurology in Clinical Practice; Lippincott Illustrated Reviews: Pharmacology
Management of hyperkalemia and hyokalemia (Pharmacology)
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hyperkalemia" AND "treatment" AND "management"
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.