Explain me about hepatitis in detail
Reading File
Finding Sources
Searching PubMed
"hepatitis"[MeSH Terms] AND ("treatment" OR "management")
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative sources. Let me compile the full detailed response.
Hepatitis - A Comprehensive Overview
Hepatitis means acute or chronic inflammation of the liver parenchyma. While it most commonly results from viral infection or toxic/alcohol exposure, other causes include bacterial, fungal, or parasitic infection; genetic disorders; autoimmune disease; and hepatotoxic medications (isoniazid, acetaminophen, and others). - Rosen's Emergency Medicine, 10e
1. Etiology and Classification
Viral Hepatitis (most common)
The five classic hepatotropic viruses are labeled A through E. Additional viral causes include Epstein-Barr virus, cytomegalovirus, and herpesvirus.
Non-viral Causes
- Toxic/Drug-induced: acetaminophen overdose (treat with N-acetylcysteine immediately), isoniazid, alternative supplements
- Alcoholic hepatitis: direct toxicity plus metabolic damage
- Autoimmune hepatitis: immune-mediated hepatocyte destruction
- Metabolic: non-alcoholic fatty liver disease (NAFLD/MASH), Wilson's disease, hemochromatosis
2. Clinical Presentation (General)
Patients with acute hepatitis often present with:
- Low-grade fever, fatigue, lethargy
- Anorexia, nausea, vomiting, diarrhea
- Right upper quadrant (RUQ) pain
- Arthralgias and myalgias
- Dark urine and jaundice (in more severe cases)
Alarm signs indicating severe hepatic destruction:
- Mental status changes (hepatic encephalopathy)
- Asterixis (liver flap)
- Ascites
- Prolonged prothrombin time (PT)
These patients may require hospitalization, nutritional support, specialist referral, and liver transplantation evaluation. Symptomatic improvement typically precedes normalization of liver enzymes. - Textbook of Family Medicine, 9e
3. Hepatitis A (HAV)
Virus: RNA enteroviral picornavirus (Picornaviridae family)
Transmission: Fecal-oral route - directly or through contaminated water and food. Blood transmission is exceedingly rare.
Epidemiology: HAV is endemic worldwide. Serologic evidence of previous infection exists in nearly 100% of adults in some regions. In the US, close to 50% of urban adults are seropositive for HAV antibody. Travel to endemic areas is the most common risk factor in persons over 15 years old.

Worldwide distribution of hepatitis A. - Rosen's Emergency Medicine
Incubation period: 15-45 days (typically 30 days), with viremia most prominent before symptom onset.
Clinical course: Symptoms occur between 2-6 weeks after infection. Up to 70% of infected children are asymptomatic. The disease is self-limiting and does NOT cause chronic infection.
Diagnosis:
- Acute: IgM anti-HAV antibody (positive in acute infection)
- Past infection/immunity: IgG anti-HAV antibody
Treatment: Supportive care. No antiviral therapy required.
Prevention:
- Formalin-inactivated HAV vaccine (licensed in the US in 1995) - recommended for all children over 1 year, international travelers, men who have sex with men, and drug users
- Post-exposure: immune globulin (IG) within 1-2 weeks of exposure provides ~90% passive protection
- Hygiene: handwashing, 0.5% sodium hypochlorite disinfectant
- HAV vaccine produces more enduring immunity and should replace IG use. - Jawetz Medical Microbiology, 28e
4. Hepatitis B (HBV)
Virus: DNA virus (Hepadnaviridae), partially double-stranded, with a lipid envelope bearing the surface antigen (HBsAg)
Transmission: Parenteral (blood, needlestick), sexual contact, and vertical (mother-to-child) transmission
Epidemiology:
- HBV vertical transmission accounts for approximately 50% of chronic infections worldwide
- Risk of chronic infection after neonatal exposure (without prophylaxis): 85-95%
- Women seropositive for both HBsAg AND HBeAg have ~90% vertical transmission rate without neonatal prophylaxis
- In the US, universal vaccination programs have significantly reduced transmission
Serologic Markers (key for diagnosis):
| Marker | Abbreviation | Meaning |
|---|---|---|
| Hepatitis B surface antigen | HBsAg | Present in acute or chronic infection |
| IgM antibody to HBcAg | HBcAb-IgM | Indicates acute HBV infection |
| HBeAg | HBeAg | Active infection, high infectivity |
| Antibody to HBsAg | HBsAb | Immunity (past infection or vaccination) |
| Antibody to HBcAg | HBcAb (total) | Past or current infection |
| Antibody to HBeAg | HBeAb | Resolving infection, decreased infectivity |
From: Rosen's Emergency Medicine
Clinical course:
- Most adults clear the infection spontaneously (~90% of immunocompetent adults)
- Risk of chronicity is inversely related to age at infection: neonates 85-95%, children 25-50%, adults ~5%
- Chronic infection can progress to cirrhosis, hepatocellular carcinoma (HCC)
Diagnosis: HBsAg + IgM anti-HBcAg = acute infection. HBcAb alone = past infection (HBsAg cleared). Anti-HBsAg alone = vaccination-induced immunity.
Treatment of Chronic HBV:
- Nucleos(t)ide analogues (NAs): entecavir, tenofovir disoproxil fumarate (TDF), tenofovir alafenamide (TAF) - first-line
- Pegylated interferon-alfa (PEG-IFN-α): finite 48-week course; limited by side effects
- The 2025 AGA guideline also provides updated protocols for prevention of HBV reactivation in at-risk individuals (PMID: 39863345)
- A 2025 systematic review (PMID: 40528088) shows new antiviral regimens are achieving functional cure (HBsAg loss) in a subset of patients
Prevention:
- Recombinant HBV vaccine is universally recommended for all infants; part of routine immunization schedule
- Post-exposure prophylaxis: Hepatitis B immunoglobulin (HBIG) + vaccine series (for unvaccinated individuals)
- Screen all pregnant women for HBsAg at the start of prenatal care
5. Hepatitis C (HCV)
Virus: Single-stranded RNA virus (Flaviviridae, genus Hepacivirus), 6 genotypes (1-6)
Transmission: Primarily parenteral (IV drug use, needle sharing, blood transfusion pre-1992 screening). Sexual and vertical transmission less efficient than HBV. Approximately 5-6% of infants born to HCV-positive mothers are infected.
Epidemiology: Increasingly affecting women of childbearing age. In the US, 0.24% of live births in 2020 were to women with reported HCV infection.
Clinical course:
- Most acute infections (~80%) are asymptomatic
- ~55-85% progress to chronic infection (compared to ~5% for HBV in adults)
- Chronic infection can lead to cirrhosis over 20-30 years, and HCC
Diagnosis:
- Screening: Anti-HCV antibody (ELISA)
- Confirmation: HCV RNA by PCR (most sensitive, detectable soon after infection; also used in chronic disease)
- Due to delayed antibody appearance, early diagnosis depends on history and excluding other causes
Treatment:
- Direct-acting antivirals (DAAs): highly effective (>95% sustained virologic response / SVR)
- Sofosbuvir-based regimens, glecaprevir/pibrentasvir, ledipasvir/sofosbuvir
- Treatment duration: 8-12 weeks typically
- No vaccine currently available for HCV
- Immunoglobulin and post-exposure prophylaxis are not effective; occupational health follow-up for baseline and follow-up testing is recommended
6. Hepatitis D (HDV) - Delta Hepatitis
Virus: Small, icosahedral, single-stranded circular (-) RNA virus (Deltaviridae). Has its own delta capsid antigen but requires HBsAg for its envelope - it is an obligate satellite of HBV.
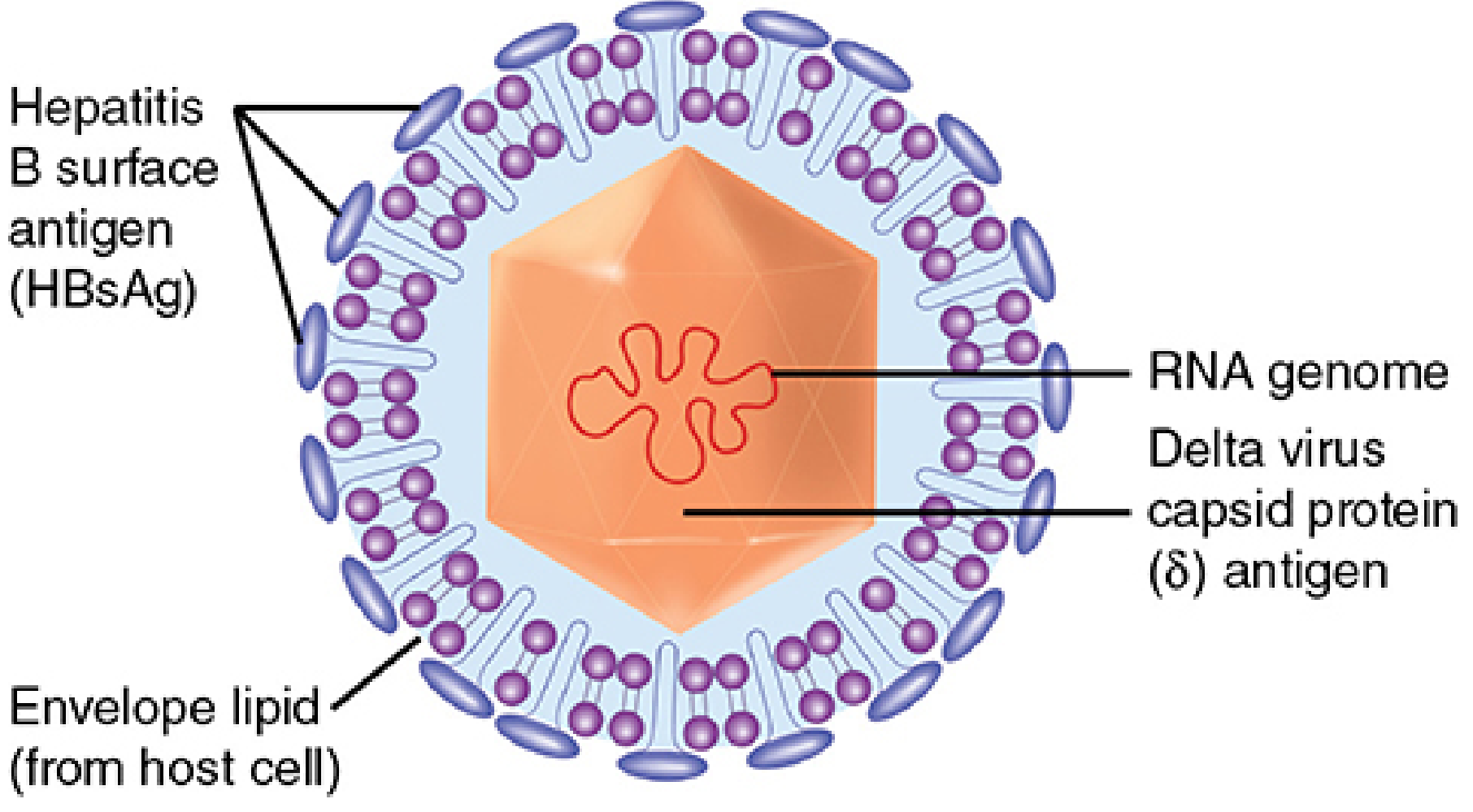
Structure of HDV - note the HBsAg on the envelope. HDV only assembles when HBsAg is present. - Sherris Medical Microbiology, 8e
Key features:
- HDV is only found in HBV-infected persons
- Uses host cell RNA polymerase (not its own) to replicate in the nucleus - unique for an RNA virus
- Transmission: same routes as HBV (parenteral, sexual)
- Greatest risk: injection drug users who are HBV-positive
Two patterns of infection:
- Co-infection: HDV + HBV acute infection simultaneously - usually self-limiting but can cause fulminant hepatitis
- Superinfection: HDV infects a chronic HBV carrier - higher risk of severe/fulminant hepatitis and worse prognosis
Prevention: HBV vaccination also prevents HDV (since HDV cannot exist without HBsAg). - Sherris Medical Microbiology, 8e
7. Hepatitis E (HEV)
Virus: Non-enveloped, single-stranded positive-sense RNA virus (Hepeviridae)
Transmission: Fecal-oral route (similar to HAV), mainly through contaminated water
Clinical course:
- Generally self-limiting
- Exception: pregnant women - HEV carries a disproportionately high mortality rate (up to 25%) in pregnant women, especially in the third trimester. This makes HEV uniquely dangerous in pregnancy.
- No progression to chronic hepatitis in immunocompetent patients (except HEV genotype 3/4 in immunosuppressed individuals)
Diagnosis: Anti-HEV IgM (acute); HEV RNA by PCR
Treatment: Supportive; ribavirin can be used in chronic HEV (immunocompromised patients)
8. Comparative Summary Table
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Genome | (+)ssRNA | dsDNA (partial) | (+)ssRNA | (-)ssRNA circular | (+)ssRNA |
| Family | Picornaviridae | Hepadnaviridae | Flaviviridae | Deltaviridae | Hepeviridae |
| Route | Fecal-oral | Parenteral/sexual/vertical | Parenteral | Parenteral/sexual | Fecal-oral |
| Incubation | 15-45 days | 45-180 days | 14-180 days | 14-56 days (coinfection) | 15-60 days |
| Chronicity | Never | ~5% (adults) | 55-85% | Higher (superinfection) | Rarely |
| HCC risk | No | Yes | Yes | Yes (via HBV) | No |
| Vaccine | Yes | Yes | No | Prevented by HBV vaccine | Yes (China only) |
9. Laboratory Findings
In all forms of acute hepatitis:
- Serum transaminases (AST, ALT) - greatly elevated (ALT typically > AST in viral hepatitis)
- Serum bilirubin - elevated
- Alkaline phosphatase (ALP) - elevated
- PT/INR - prolonged in severe disease (marker of synthetic function loss)
The specific values have poor prognostic value individually; clinical features matter more. - Textbook of Family Medicine, 9e
10. Hepatitis in Pregnancy
Special considerations (from Goldman-Cecil Medicine):
- Hepatitis B vertical transmission responsible for ~50% of chronic infections worldwide
- Risk of chronic HBV if acquired perinatally: 85-95% with 25-30% lifetime risk of serious liver disease
- HBsAg is measured at the start of every prenatal visit
- Indications for antiviral therapy in pregnancy are the same as in non-pregnant patients
- Women with high viral load (HBV DNA >200,000 IU/mL) in the third trimester should receive antiviral prophylaxis (tenofovir) to reduce vertical transmission
11. Imaging
On ultrasound:
- Acute hepatitis: liver enlargement, diffuse decrease in parenchymal echogenicity, echogenic portal triads ("starry sky" pattern - though nonspecific)
- Chronic hepatitis: may show coarsened echotexture, increased parenchymal echogenicity, periportal lymphadenopathy
- Gallbladder wall thickening can be present in acute hepatitis
- Yamada's Textbook of Gastroenterology notes that chronic hepatitis can appear completely normal on ultrasound in many cases
12. Recent Evidence Updates (2025)
- A 2025 systematic review and meta-analysis (PMID: 40528088) found that new antiviral regimens including combined NA + PEG-IFN, or novel capsid assembly modulators, are achieving functional cure (HBsAg clearance) in a meaningful subset of chronic HBV patients - a development not possible with older standard-of-care monotherapy.
- A 2025 AGA clinical practice guideline (PMID: 39863345) provides updated recommendations on preventing HBV reactivation in at-risk individuals (those on immunosuppressive therapy, biologics, chemotherapy).
- A 2025 systematic review (PMID: 39874480) on corticosteroids in alcohol-associated hepatitis found that the evidence for steroid benefit remains uncertain - cautioning against routine use in severe alcoholic hepatitis.
Sources: Rosen's Emergency Medicine (10e), Textbook of Family Medicine (9e), Goldman-Cecil Medicine, Jawetz Medical Microbiology (28e), Sherris Medical Microbiology (8e), Yamada's Textbook of Gastroenterology (7e), Washington Manual of Medical Therapeutics
Type Definition Causes Clinical features,sign and symptoms Investigation with expected findings and why we do that to asses what Complications Prevention Management(pharmacology and non pharmacology ) Pathophysiology Diagnostic criteria Ddx with points of favour and against
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
HEPATITIS - Complete Clinical Reference
1. DEFINITION
Hepatitis is defined as acute or chronic inflammation of the hepatic parenchyma (liver cells - hepatocytes). The term encompasses a spectrum from asymptomatic enzyme elevation to fulminant liver failure. Acute hepatitis refers to injury lasting less than 6 months; chronic hepatitis is inflammation persisting for 6 months or more, or signs of chronic liver disease in the presence of persistently elevated cytosolic enzymes. - Tietz Textbook of Laboratory Medicine, 7e; Rosen's Emergency Medicine
2. TYPES
A. By Duration
| Type | Duration | Key Feature |
|---|---|---|
| Acute | < 6 months | Self-limiting in most; may progress to fulminant failure |
| Chronic | ≥ 6 months | Risk of cirrhosis and HCC |
| Fulminant | Acute + encephalopathy within 8 weeks | High mortality without transplant |
B. By Etiology
| Type | Cause |
|---|---|
| Hepatitis A (HAV) | RNA picornavirus, fecal-oral |
| Hepatitis B (HBV) | DNA hepadnavirus, parenteral/sexual/vertical |
| Hepatitis C (HCV) | RNA flavivirus, parenteral |
| Hepatitis D (HDV) | Defective RNA virus, requires HBV |
| Hepatitis E (HEV) | RNA hepeviridae, fecal-oral |
| Alcoholic hepatitis | Ethanol toxicity |
| Drug-induced (DILI) | Direct toxicity or idiosyncratic |
| Autoimmune hepatitis | Immune-mediated hepatocyte attack |
| Non-alcoholic steatohepatitis (NASH/MASH) | Metabolic syndrome |
| Ischemic ("shock liver") | Hypoperfusion/circulatory failure |
3. CAUSES (Detailed)
Infectious
- HAV: RNA enteroviral picornavirus. Spread by fecal-oral route through contaminated food/water. Sporadic or epidemic (restaurants, shellfish). Incubation 15-45 days.
- HBV: Small partially double-stranded DNA virus (Hepadnaviridae). Transmitted parenterally (blood, needlestick), sexually, and vertically (mother-to-child). Incubation 45-180 days.
- HCV: Single-stranded (+) RNA virus (Flaviviridae), 6 genotypes. Primarily parenteral (IVDU, pre-1992 transfusions). Incubation 14-180 days. ~80% asymptomatic acutely.
- HDV (Delta hepatitis): Small icosahedral (-) ssRNA circular virus (Deltaviridae). Requires HBsAg for its lipid envelope - only infects HBV carriers. Parenteral/sexual. Greatest risk in IV drug users.
- HEV: Non-enveloped (+) ssRNA (Hepeviridae). Fecal-oral, contaminated water, endemic in developing countries. Uniquely severe in pregnancy (up to 25% mortality in third trimester).
- Other viruses: EBV (mononucleosis), CMV, HSV, adenovirus, coxsackievirus, COVID-19
Toxic/Drug-Induced
- Acetaminophen overdose - dose-dependent direct hepatotoxin (most common cause of acute liver failure in the West)
- Alcohol - direct hepatotoxicity + metabolic disruption
- Isoniazid, rifampicin, pyrazinamide (anti-TB)
- Statins, valproate, amiodarone, methotrexate
- Herbal supplements (e.g., kava, comfrey)
- Amanita phalloides mushroom poisoning
- Carbon tetrachloride (occupational exposure)
Autoimmune
- Type 1: ANA + anti-smooth muscle antibody (SMA) - predominantly young women
- Type 2: Anti-LKM1 (liver-kidney microsome) antibody - children, Mediterranean populations
Metabolic
- NAFLD/NASH: Fatty change progressing to steatohepatitis
- Wilson's disease: Copper accumulation - "the great imitator"
- Alpha-1 antitrypsin deficiency
- Hemochromatosis
Ischemic / Vascular
- Shock liver (ischemic hepatitis): Massive AST/ALT elevation (>20x ULN) with rapid normalization after hemodynamic restoration; bilirubin paradoxically only mildly elevated
4. CLINICAL FEATURES, SIGNS AND SYMPTOMS
Prodromal Phase (1-2 weeks before jaundice)
- Fatigue, malaise, lethargy
- Anorexia, nausea, vomiting
- Low-grade fever
- Arthralgias and myalgias
- Right upper quadrant (RUQ) discomfort
- Headache, photophobia (rare)
Icteric Phase (jaundice appears)
- Jaundice (scleral icterus first, then skin) - lasts 2-12 weeks
- Dark urine (bilirubinuria - conjugated bilirubin spills into urine)
- Pale/clay-coloured stools (reduced bilirubin reaching gut)
- Pruritus (bile salt deposition in skin)
- Hepatomegaly, liver tenderness on palpation
- Splenomegaly (~25% of cases)
- Resolution phase: Symptoms improve before liver enzymes normalize
Signs of Severe/Chronic Disease
- Hepatic encephalopathy: confusion, asterixis (liver flap), somnolence
- Ascites (portal hypertension, hypoalbuminaemia)
- Spider angiomata, palmar erythema (oestrogen excess in chronic disease)
- Caput medusae (dilated periumbilical veins)
- Splenomegaly with thrombocytopenia (hypersplenism)
- Coagulopathy (prolonged PT/INR)
- Gynecomastia (chronic liver disease)
- Dupuytren's contracture (alcoholic liver disease)
Extrahepatic Manifestations (especially HCV)
- Cryoglobulinemia: purpura, arthralgias, Raynaud phenomenon, peripheral neuropathy
- Membranoproliferative glomerulonephritis / cryoglobulinemic nephropathy
- Sicca syndrome, vasculitis
- HBV: polyarteritis nodosa, membranous nephropathy, serum sickness-like prodrome
5. INVESTIGATIONS - WITH EXPECTED FINDINGS AND RATIONALE
A. Liver Function Tests (LFTs)
| Test | Expected Finding | Why We Do It / What It Assesses |
|---|---|---|
| ALT (Alanine Aminotransferase) | 8-50× ULN in acute viral hepatitis; >300 U/L (>90% sensitivity for acute hepatitis) | Most specific marker of hepatocyte injury; cytosolic enzyme released when hepatocytes are damaged. ALT > AST in viral hepatitis |
| AST (Aspartate Aminotransferase) | Elevated, but usually less than ALT in viral hepatitis | Also present in muscle, heart; less specific. AST/ALT >2:1 suggests alcoholic hepatitis |
| ALP (Alkaline Phosphatase) | Mildly elevated (<3× ULN in 90% of acute hepatitis) | Elevated mainly in cholestatic disease; modest rise distinguishes hepatitis from biliary obstruction |
| GGT (Gamma-glutamyl transferase) | Elevated; disproportionately raised in alcohol use | Sensitive but non-specific; confirms hepatic origin of elevated ALP; marker of alcohol intake |
| Serum Bilirubin (total, direct, indirect) | Elevated total; predominantly direct (conjugated) | Assesses degree of jaundice; direct hyperbilirubinemia indicates conjugation is intact but secretion impaired |
| Albumin | Low in chronic/severe disease (only decreased when >80% liver function lost) | Marker of hepatic synthetic function; long half-life (~21 days) so falls late |
| Prothrombin Time (PT)/INR | Prolonged in severe disease | Liver synthesizes all clotting factors except VIII; PT is the most sensitive marker of acute synthetic failure. Best prognostic indicator |
| Serum Ammonia | Elevated in hepatic encephalopathy | Liver converts ammonia to urea; rises when >80% liver tissue destroyed |
| LDH | Moderately elevated | Additional cytosolic enzyme; less specific |
Key Pattern in Acute Hepatitis: ALT and AST markedly elevated (8-50×), ALP mildly elevated (<3×), bilirubin variable. Enzyme elevations peak before peak bilirubin. ALT typically remains elevated longer than AST due to its longer half-life. - Tietz Textbook of Laboratory Medicine, 7e
B. Serological Markers (to identify the specific cause)
| Test | When Positive | What It Means |
|---|---|---|
| IgM anti-HAV | Acute HAV | Confirms acute hepatitis A infection |
| IgG anti-HAV | Past infection or vaccination | Immunity to HAV |
| HBsAg | Acute or chronic HBV | Surface antigen; present if actively infected; >6 months = chronic |
| IgM anti-HBcAg | Acute HBV | Best marker of acute HBV infection; distinguishes acute from chronic |
| HBeAg | Active HBV replication | High infectivity; present in active replication |
| HBeAb | Seroconversion | Marker of resolving/resolved replication; low infectivity |
| Anti-HBsAg | Recovery or vaccination | Protective immunity |
| HBV DNA (quantitative PCR) | Detectable in replicating HBV | Measures viral load; guides treatment decisions |
| Anti-HCV (ELISA) | Screening for HCV | Does NOT distinguish acute from past; may be negative early in acute infection |
| HCV RNA (PCR) | Acute or chronic HCV | Definitive confirmation; detectable weeks before antibody; also guides treatment |
| HCV genotype | Chronic HCV | Determines treatment regimen and duration |
| Anti-HDV | HDV infection | HBsAg must also be present |
| IgM anti-HEV / HEV RNA | Acute HEV | Confirm hepatitis E |
C. Additional Investigations
| Test | Findings | Purpose |
|---|---|---|
| FBC (Full Blood Count) | Leukopenia + lymphocytosis (viral); thrombocytopenia (chronic/hypersplenism) | Assess haematological impact; leucocytosis in alcoholic hepatitis |
| Glucose | Hypoglycemia (alcoholic hepatitis - impaired gluconeogenesis + depleted glycogen) | Detect metabolic complications |
| Coagulation screen (PT, APTT) | Prolonged PT indicates severe disease | Severity assessment and prognostication |
| Renal function (U&E, creatinine) | Rising creatinine + liver failure = hepatorenal syndrome | Detect renal complications |
| ANA, anti-SMA, anti-LKM1 | Positive in autoimmune hepatitis | Rule out autoimmune cause; essential when viral serology negative |
| IgG level | Raised in autoimmune hepatitis | Part of diagnostic scoring for AIH |
| Ceruloplasmin, urinary copper | Low ceruloplasmin in Wilson's disease | Rule out Wilson's, especially in young patients with low ALP |
| Alpha-1 antitrypsin level | Low in AAT deficiency | Metabolic cause of chronic hepatitis |
| Ferritin, transferrin saturation | Elevated in haemochromatosis | Rule out iron overload |
| Ultrasound abdomen | Hepatomegaly, decreased echogenicity ("starry sky"), gallbladder wall thickening (acute); coarsened echotexture, periportal lymphadenopathy (chronic) | First-line imaging; non-invasive assessment of liver parenchyma, biliary tree, portal circulation |
| CT / MRI liver | Periportal enhancement on contrast MRI (late arterial phase 18-22s) is most sensitive for acute hepatitis; periportal edema on T2 | MRI is most sensitive imaging modality for acute hepatitis |
| Liver biopsy | Interface hepatitis, lobular inflammation, hepatocyte necrosis, fibrosis staging | Gold standard for grading inflammation and staging fibrosis; guides prognosis and treatment decisions |
| Ultrasound elastography (Fibroscan) | Increased liver stiffness correlates with fibrosis | Non-invasive alternative to biopsy for assessing fibrosis stage in chronic hepatitis |
| Upper GI endoscopy | Oesophageal varices | Indicated if portal hypertension suspected; screens for variceal bleeding |
6. PATHOPHYSIOLOGY
Viral Hepatitis (Immune-Mediated Mechanism - HBV Model)
HBV is NOT directly cytopathic. Liver damage is mediated by the host immune response:
- Viral entry: HBV enters hepatocytes via sodium-taurocholate cotransporting polypeptide (NTCP) receptor using HBsAg
- Viral replication: HBV DNA replicates in the nucleus; viral proteins (HBsAg, HBcAg, HBeAg) are expressed on the hepatocyte surface
- Immune recognition: CD8+ cytotoxic T lymphocytes (CTLs) recognize viral antigens on MHC class I molecules on hepatocyte surface
- Hepatocyte destruction: CTLs attack infected hepatocytes causing necrosis and apoptosis (the primary mechanism of liver damage)
- Innate immune response: NK cells, interferon release also contribute
- Wound-healing response: Repeated hepatocyte injury triggers activation of hepatic stellate cells (Ito cells) → collagen deposition → fibrosis
- Chronic inflammation → progressive fibrosis → cirrhosis → portal hypertension + risk of hepatocellular carcinoma (HCC)
Phases of Chronic HBV (Immune Staging):
| Phase | HBeAg | HBV DNA | ALT | Meaning |
|---|---|---|---|---|
| Immune tolerant | + | Very high | Normal | Virus replicates freely; no immune attack |
| Immune reactive | + → - | Falling | Elevated | Immune system fighting; active hepatitis |
| Immune control (inactive carrier) | - | Low/undetectable | Normal | Virus suppressed; low risk |
| HBeAg-negative reactivation | - | Rising | Elevated | Precore mutant HBV; active disease despite HBeAg negativity |
HAV / HEV Pathophysiology (Direct Injury)
- These enteric viruses have some degree of direct cytopathic effect in addition to immune-mediated damage
- Inflammation is generally self-limiting as virus is cleared; no chronic state
Alcoholic Hepatitis
- Alcohol metabolism produces acetaldehyde and ROS (reactive oxygen species) → direct hepatocyte membrane damage
- Lipid peroxidation → steatosis
- Gut dysbiosis + increased gut permeability → endotoxin (LPS) enters portal circulation → activates Kupffer cells → pro-inflammatory cytokine release (TNF-α, IL-1β, IL-6, IL-8)
- Mallory-Denk bodies (abnormal intermediate filament aggregates) form in hepatocytes
- Histology: steatosis, lobular neutrophilic infiltration, hepatocyte ballooning, Mallory-Denk bodies, pericellular fibrosis
- AST predominates over ALT (>2:1 ratio) because: alcohol depletes pyridoxal phosphate (ALT requires more of it than AST), and alcohol-damaged mitochondria release mitochondrial AST
Drug-Induced Hepatitis
- Type 1 - Direct/dose-dependent (e.g. acetaminophen): overdose depletes glutathione stores → N-acetyl-p-benzoquinone imine (NAPQI) accumulates → oxidative hepatocyte destruction → Zone 3 centrilobular necrosis (highest CYP2E1 activity)
- Type 2 - Idiosyncratic: unpredictable, immune-mediated, dose-independent (e.g. isoniazid, halothane); mimics viral hepatitis
7. DIAGNOSTIC CRITERIA
A. Acute Viral Hepatitis (General)
No formal scoring system; diagnosis is based on:
- ALT/AST >8-50× ULN (sensitivity >90% if ALT >300 or AST >200)
- ALP <3× ULN (distinguishes from cholestatic disease)
- Positive specific serology (see above)
B. Chronic Hepatitis
- Duration of inflammation ≥6 months (clinical or biochemical)
- Confirmed etiology by serology/autoantibodies/histology
- Biopsy: graded (inflammatory activity) and staged (fibrosis)
C. Autoimmune Hepatitis (AIH) - Simplified Scoring System
(International Autoimmune Hepatitis Group - Simplified Criteria)
| Variable | Points |
|---|---|
| ANA or SMA ≥ 1:40 | 1 |
| ANA or SMA ≥ 1:80, OR anti-LKM ≥ 1:40, OR anti-SLA positive | 2 |
| IgG > ULN | 1 |
| IgG > 1.1 × ULN | 2 |
| Histology compatible with AIH | 1 |
| Histology typical for AIH | 2 |
| Absence of viral hepatitis | 2 |
- ≥6 points = probable AIH
- ≥7 points = definite AIH
- Max autoantibody points = 2. - Symptom to Diagnosis: An Evidence-Based Guide, 4e
D. Alcoholic Hepatitis Severity - Maddrey Discriminant Function (MDF)
MDF = 4.6 × (PT_patient - PT_control) + serum bilirubin (mg/dL)
- MDF > 32 = severe disease → consider corticosteroids
- MELD score >20 also indicates severe disease
E. Chronic HBV - Indications for Treatment
HBsAg positive >6 months, AND one of:
- HBV DNA >2,000 IU/mL (HBeAg-negative) or >20,000 IU/mL (HBeAg-positive)
- ALT >2× ULN
- Liver biopsy showing moderate/severe inflammation or significant fibrosis
- Decompensated cirrhosis (any HBV DNA level) - Goldman-Cecil Medicine
8. COMPLICATIONS
A. Acute Complications
| Complication | Details |
|---|---|
| Fulminant hepatic failure | Massive hepatic necrosis; hepatic encephalopathy within 8 weeks of onset; high mortality. More common with HBV (especially coinfection with HDV), HEV (in pregnancy), Wilson's disease, drug-induced |
| Cholestatic hepatitis | Prolonged jaundice without significant parenchymal damage; more common in HAV |
| Aplastic anaemia | Rare; follows HAV |
| Relapsing hepatitis | HAV can relapse weeks after apparent recovery |
B. Chronic Complications
| Complication | Mechanism | Details |
|---|---|---|
| Cirrhosis | Chronic inflammation → fibrosis | Irreversible; leads to portal hypertension. Risk: HBV ~25-30% lifetime; HCV ~20-30% over 20-30 years |
| Portal hypertension | Increased resistance to portal flow in cirrhosis | Leads to varices, splenomegaly, ascites |
| Oesophageal varices + bleeding | Portal hypertension → portosystemic shunting | 10-15% mortality per bleeding episode |
| Ascites | Portal hypertension + hypoalbuminaemia | Fluid accumulation in peritoneal cavity |
| Hepatic encephalopathy | Elevated ammonia + other toxins affect cerebral metabolism | Confusion → stupor → coma; precipitated by GI bleeding, infection, hypokalemia, dehydration |
| Spontaneous bacterial peritonitis (SBP) | Translocation of gut bacteria into ascitic fluid | Fever + abdominal pain in cirrhotic patients; neutrophils >250/mm³ in ascitic fluid |
| Hepatorenal syndrome | Vasoconstriction of renal arteries in severe liver disease | Rising creatinine + liver failure; high mortality |
| Hepatocellular carcinoma (HCC) | Chronic inflammation → regeneration cycles → oncogenic mutations (HBV X protein also directly mutagenic) | HBV risk: 4× higher in men; HCC can occur even without cirrhosis in HBV; screen with 6-monthly USS + AFP |
| Cryoglobulinemia (HCV) | HCV drives B-cell clonal expansion | Purpura, arthritis, glomerulonephritis, neuropathy |
| Coagulopathy | Impaired clotting factor synthesis | Prolonged PT/INR; bleeding tendency |
9. PREVENTION
A. Vaccination
| Hepatitis | Vaccine | Schedule |
|---|---|---|
| Hepatitis A | Formalin-inactivated (e.g. Havrix, Vaqta) | 2 doses, 6-12 months apart; all children >1 year; high-risk adults |
| Hepatitis B | Recombinant HBsAg (e.g. Engerix-B, Recombivax) | 3 doses (0, 1, 6 months); universal infant vaccination; all unvaccinated individuals |
| Hepatitis D | Prevented by HBV vaccination | No separate HDV vaccine needed |
| Hepatitis E | HEV 239 (Hecolin) | Available in China; not widely approved elsewhere |
| Hepatitis C | None | Prevention relies entirely on harm reduction |
B. Post-Exposure Prophylaxis (PEP)
| Exposure | Prophylaxis |
|---|---|
| HAV exposure | Immune globulin (IG) within 1-2 weeks OR HAV vaccine (preferred) |
| HBV exposure (unvaccinated) | HBIG 0.06 mL/kg IM within 96 hours + start HBV vaccine series |
| HBV exposure (vaccinated, known responder with anti-HBs ≥10 mIU/mL) | No treatment needed |
| HBV exposure (vaccinated, nonresponder after 1 complete series) | HBIG × 1 dose + repeat vaccine series |
| HCV exposure | No effective PEP; baseline testing + occupational health follow-up |
C. Harm Reduction Strategies
- Safe sex education, condom use (HBV, HCV)
- Needle exchange programs and opioid substitution therapy (HBV, HCV)
- Safe blood supply screening (blood banks screen for HBsAg, anti-HCV)
- Universal precautions in healthcare settings (gloves, masks, no needle recapping)
- Antenatal screening for HBsAg in all pregnant women
- Neonatal HBIG + HBV vaccine for infants of HBsAg-positive mothers (within 12 hours of birth)
- Food hygiene, clean water supply, handwashing (HAV, HEV)
10. MANAGEMENT
A. Non-Pharmacological
| Measure | Rationale |
|---|---|
| Rest | Reduce metabolic demands on injured liver |
| Adequate nutrition | Liver regeneration requires adequate caloric and protein intake; high-carb, low-fat diet in acute phase |
| Abstinence from alcohol | Alcohol accelerates fibrosis progression; steatosis can reverse within 2 weeks of cessation |
| Avoid hepatotoxic drugs | NSAIDs, paracetamol (standard doses acceptable in viral hepatitis; avoid in severe disease), statins |
| Contact isolation precautions | HAV/HEV: enteric precautions; HBV/HCV: standard blood/body fluid precautions |
| Activity restriction | Gradual return to activity; symptomatic improvement precedes biochemical normalisation |
| Management of complications: | |
| - Variceal bleeding | Banding, propranolol for prophylaxis |
| - Ascites | Salt restriction + spironolactone ± furosemide; therapeutic paracentesis (+ albumin 8g/L removed if >5L) |
| - Encephalopathy | Lactulose, treat precipitants, dietary protein adjustment |
| - HCC surveillance | 6-monthly ultrasound + AFP in all cirrhotic patients and HBV carriers |
| Liver transplantation | Indicated for decompensated cirrhosis, acute liver failure, or HCC within Milan criteria |
B. Pharmacological
Hepatitis B
| Medication | Class | Dose | Duration | Notes |
|---|---|---|---|---|
| Tenofovir disoproxil fumarate (TDF) | Nucleotide analogue | 300 mg/day PO | Long-term/indefinite | Preferred first-line; effective against lamivudine-resistant HBV; monitor renal function and bone density |
| Tenofovir alafenamide (TAF) | Nucleotide analogue | 25 mg/day PO | Long-term/indefinite | Better renal/bone safety profile than TDF; preferred in renal impairment |
| Entecavir | Nucleoside analogue | 0.5 mg/day PO (naïve); 1 mg/day (lamivudine-resistant) | Long-term/indefinite | Preferred first-line; high barrier to resistance |
| Pegylated interferon-alfa-2a | Immunomodulator | 180 μg SQ weekly | 48 weeks | Finite course; induces immune-mediated viral clearance; better seroconversion rates but poor tolerability (flu-like symptoms, depression, cytopenia) |
| Lamivudine | Nucleoside analogue | 100 mg/day | No longer preferred | High resistance rates with long-term use |
| Adefovir | Nucleotide analogue | 10 mg/day | No longer preferred | Replaced by TDF |
Treatment endpoints:
- HBeAg-positive: Continue until HBeAg seroconversion + undetectable HBV DNA, then ≥6 more months
- HBeAg-negative: At least 1 year; often lifelong
- Decompensated cirrhosis: Lifelong
Hepatitis C - Direct-Acting Antivirals (DAAs)
| Regimen | Target | Genotypes | Duration | SVR Rate |
|---|---|---|---|---|
| Sofosbuvir/Velpatasvir (Epclusa) | NS5B pol + NS5A | Pan-genotypic (1-6) | 12 weeks | >95% |
| Glecaprevir/Pibrentasvir (Mavyret) | NS3/4A protease + NS5A | Pan-genotypic | 8 weeks (treatment-naïve, no cirrhosis) | >97% |
| Ledipasvir/Sofosbuvir (Harvoni) | NS5A + NS5B | Genotype 1, 4, 5, 6 | 8-12 weeks | >95% |
SVR (Sustained Virologic Response = undetectable HCV RNA 12 weeks post-treatment) = functional cure
Hepatitis A and E
- Supportive care only - no antiviral therapy available or required
- HAV: self-limiting; complete recovery expected
- HEV immunocompromised patients: Ribavirin (off-label, 12-24 weeks) for chronic HEV
Autoimmune Hepatitis
- Induction: Prednisone 40-60 mg/day alone, OR Prednisone 30 mg/day + Azathioprine 50 mg/day
- Maintenance: Azathioprine 1-2 mg/kg/day (most patients need lifelong therapy)
- Remission achieved in ~85% of patients
- Alternative agents (MMF, tacrolimus, cyclosporine) for non-responders/intolerant
Alcoholic Hepatitis (MDF >32 / MELD >20)
- Prednisone 40 mg/day PO or methylprednisolone 32 mg IV daily for 28 days (if no contraindication)
- Contraindications to steroids: active GI bleeding, sepsis, renal failure
- Pentoxifylline 400 mg TID (alternative if steroids contraindicated - inhibits TNF-α)
- Note: A 2025 systematic review (PMID: 39874480) challenges routine steroid use, finding uncertain benefit across RCTs
Fulminant Hepatic Failure
- N-acetylcysteine (NAC): Treatment of choice for acetaminophen-induced ALF; also benefits non-acetaminophen ALF
- Acute HBV (severe): Entecavir 0.5 mg/day or TDF 300 mg/day
- Lactulose for encephalopathy (15-30 mL TID/QID to produce 2-3 soft stools/day)
- Liver transplantation for acute liver failure unresponsive to medical therapy
11. DIFFERENTIAL DIAGNOSIS (DDx) WITH POINTS FOR AND AGAINST
| Condition | Points IN FAVOUR | Points AGAINST |
|---|---|---|
| Viral Hepatitis A | Fecal-oral exposure; raw shellfish/restaurant food; travel to endemic area; prodrome + jaundice; tender hepatomegaly; IgM anti-HAV positive | No chronic course; blood/sexual exposure history; HBsAg or anti-HCV positive |
| Viral Hepatitis B | Parenteral/sexual/perinatal exposure; HBsAg positive; IgM anti-HBcAg positive; can be chronic; significant serum sickness prodrome (urticaria, arthritis) | HAV exposure, fecal-oral route; IgM anti-HAV positive |
| Viral Hepatitis C | IV drug use; transfusion pre-1992; tattoo/piercing; anti-HCV positive; usually asymptomatic acutely | Usually no prodrome or jaundice acutely; predominantly AST/ALT pattern similar to HBV |
| Alcoholic Hepatitis | Heavy alcohol history; AST:ALT >2:1; AST rarely >300 U/L; fever + leukocytosis; tender hepatomegaly; hypoglycemia; signs of chronic liver disease (palmar erythema, spider naevi) | Viral serology positive; ALT > AST; no alcohol history; autoantibodies present |
| Autoimmune Hepatitis | Young woman; fluctuating transaminases; positive ANA/SMA/anti-LKM1; elevated IgG; other autoimmune conditions; responds to steroids | Viral serology positive; alcohol history; drug exposure; AIH scoring <6 |
| Drug-Induced Liver Injury (DILI) | Recent new drug or supplement; temporal relationship with drug initiation; eosinophilia (hypersensitivity type); improvement after drug withdrawal | All serology negative with no drug history; onset months before any new drug |
| Biliary Obstruction (choledocholithiasis, cholangitis) | ALP disproportionately elevated (>3×); RUQ colicky pain; biliary dilation on USS; fever + rigors (cholangitis); elevated amylase if pancreatitis | Predominant ALT/AST rise; no biliary dilation on imaging; prodromal viral illness |
| Ischaemic Hepatitis (Shock liver) | Recent hypotension/sepsis/cardiac failure; extremely rapid rise AND fall of transaminases (peaks within 1-3 days); bilirubin disproportionately mild for degree of transaminase rise | No haemodynamic compromise; gradual enzyme rise over days-weeks |
| Wilson's Disease | Young patient (<40 years); low ALP (paradoxically low despite hepatitis - diagnostic clue); haemolytic anaemia; Kayser-Fleischer rings on slit-lamp; low ceruloplasmin; elevated urinary copper; neuropsychiatric symptoms | Older patient; positive viral serology; normal copper studies; normal ALP |
| EBV/CMV Hepatitis | Pharyngitis + lymphadenopathy + splenomegaly; heterophile antibodies (Monospot); EBV/CMV IgM positive; younger patients; lymphocytosis with atypical lymphocytes on blood film | Hepatitis-specific serology positive; no lymphadenopathy or pharyngitis |
| NASH (Non-Alcoholic Steatohepatitis) | Metabolic syndrome (obesity, DM, hypertriglyceridemia); gradual mild elevation (1-4×); normal viral serology and autoantibodies; steatosis on ultrasound; AST/ALT usually <1 | Positive viral serology; acute presentation; significant jaundice; no metabolic risk factors |
Sources:
- Rosen's Emergency Medicine, 10e
- Tietz Textbook of Laboratory Medicine, 7e
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease
- Goldman-Cecil Medicine
- Symptom to Diagnosis: An Evidence-Based Guide, 4e
- Morgan and Mikhail's Clinical Anesthesiology, 7e
- Yamada's Textbook of Gastroenterology, 7e
- Jawetz Medical Microbiology, 28e
- Sherris Medical Microbiology, 8e
- Textbook of Family Medicine, 9e
Explained it
Asking for Preferences
I mean everything in detail
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Here is everything explained in full detail, in plain language with the underlying "why" at every step.
HEPATITIS - Everything Explained in Full Detail
SECTION 1: TYPES
Think of hepatitis as a fire in the liver. The fire can come from different sources, burn for different durations, and cause different amounts of damage. There are two main ways to classify it:
By Duration
Acute Hepatitis (< 6 months)
The liver is suddenly inflamed. This is like a short, intense fire. The body usually puts it out on its own. Most acute hepatitis resolves completely within weeks to months. However, in rare cases (especially with Hepatitis B, D, or toxic injury), the fire can become so overwhelming that massive liver cell death occurs within days - this is called fulminant hepatic failure, where the liver collapses so fast that it cannot recover.
Chronic Hepatitis (≥ 6 months)
The inflammation has been going on for more than six months. This is like smouldering embers that never fully go out. The liver keeps trying to repair itself, but each repair lays down scar tissue. Over years, scar tissue replaces normal liver tissue - this is called fibrosis, and its end stage is cirrhosis.
Fulminant Hepatic Failure
This is the most dangerous form. The liver loses so much function so rapidly that the brain becomes toxic (hepatic encephalopathy). Without a liver transplant, mortality is very high.
By Cause
| Type | Virus/Agent | Route | Chronic? | Vaccine? |
|---|---|---|---|---|
| Hepatitis A | RNA picornavirus | Fecal-oral (contaminated food/water) | Never | Yes |
| Hepatitis B | DNA hepadnavirus | Blood, sex, mother to baby | Yes (~5% adults) | Yes |
| Hepatitis C | RNA flavivirus | Mainly blood (needles) | Yes (~80%) | No |
| Hepatitis D | Defective RNA virus | Blood, sex (needs HBV) | Yes | Via HBV vaccine |
| Hepatitis E | RNA hepeviridae | Fecal-oral (contaminated water) | Rarely (only immunosuppressed) | Yes (China only) |
| Alcoholic | Ethanol + metabolites | Ingestion | Yes (if continued) | N/A |
| Drug-induced | Drugs/toxins | Ingestion/injection | Yes (some) | N/A |
| Autoimmune | Own immune system attacks liver | N/A (internal) | Yes | N/A |
| NASH/NAFLD | Fat accumulation | Metabolic syndrome | Yes | N/A |
| Ischemic | Low blood flow to liver | Shock, cardiac failure | No | N/A |
SECTION 2: DEFINITION
Hepatitis = inflammation of the hepatic parenchyma (liver cells).
To understand this, you need to understand what the liver does normally:
- It processes everything you eat, drink, and absorb
- It makes proteins (albumin, clotting factors)
- It detoxifies drugs, alcohol, and waste products
- It makes bile to digest fat
- It converts ammonia (from protein breakdown) into urea (excreted by kidneys)
- It stores glycogen (energy reserve)
When the liver cells (hepatocytes) get inflamed and die, all these functions get disrupted. The symptoms of hepatitis are simply the consequences of these functions failing.
SECTION 3: CAUSES (Deep Explanation)
Why Does HAV Cause Hepatitis?
HAV is a picornavirus (tiny RNA virus). When you swallow contaminated food/water, the virus reaches your gut → is absorbed → travels in the blood to the liver → enters hepatocytes. Once inside, it replicates and the immune system responds. HAV causes some direct cell damage, but most of the inflammation is from the immune system attacking infected hepatocytes. Since the immune system successfully clears HAV, it never becomes chronic. You develop lifelong immunity after recovery.
Why Does HBV Cause Both Acute and Chronic Hepatitis?
HBV is clever. It is a DNA virus - it enters the nucleus of hepatocytes and creates a very stable circular DNA form called cccDNA (covalently closed circular DNA). This acts like a permanent archive inside the cell. Even if antiviral drugs suppress the virus in the blood, cccDNA stays in the nucleus and can re-activate later.
Why some people clear it and others don't: Depends entirely on the immune response mounted at the time of infection:
- Neonates: Their immune system is immature and "tolerant." They cannot mount an effective T-cell response. The virus hides. Result: 85-95% become chronic.
- Adults with healthy immune systems: Strong CD8+ T-cell response clears the virus. Result: 95% clear it.
- Immunosuppressed adults: Poor response → higher chronicity rate.
Why Does HCV Almost Always Become Chronic?
HCV is an RNA virus that mutates at an extraordinary rate. Every time it replicates it creates slightly different versions of itself (called quasispecies). The immune system learns to recognize and attack one version - but by the time it does, the virus has already changed. It constantly evades immune surveillance. This is also why a vaccine has been impossible to develop - there is no single stable target to vaccinate against.
Why Does HDV Require HBV?
HDV is a defective satellite virus - it cannot complete its own lifecycle. It encodes its own internal protein (delta antigen) but it cannot make its own outer coat (envelope). To assemble itself and exit the cell to infect new hepatocytes, it steals HBsAg from HBV as its envelope. Without HBsAg, HDV cannot spread. This is why HBV vaccination also prevents HDV.
Why Is HEV Lethal in Pregnancy?
HEV normally causes a mild self-limiting illness. But during pregnancy - especially the third trimester - two factors combine dangerously:
- Hormonal immunosuppression: Pregnancy naturally dampens the immune system to protect the fetus. This allows HEV to replicate more aggressively.
- Altered liver metabolism: The liver is working harder in pregnancy due to increased metabolic demands.
The result is fulminant hepatic failure occurring in up to 25% of infected pregnant women - one of the highest disease-specific maternal mortality rates in infectious disease.
Why Does Alcohol Damage the Liver?
Alcohol (ethanol) causes a cascade of injury:
Step 1 - Metabolism of alcohol produces toxic byproducts:
- Ethanol → (Alcohol dehydrogenase) → Acetaldehyde → very toxic, binds to proteins and DNA, causes direct hepatocyte membrane damage and triggers immune response
- Acetaldehyde → (Acetaldehyde dehydrogenase) → Acetate (harmless)
- Excess NADH is produced in this process → blocks normal fat oxidation → fat accumulates in liver = steatosis (fatty liver)
Step 2 - Oxidative stress:
- The CYP2E1 pathway (another alcohol metabolism route) generates reactive oxygen species (ROS) → lipid peroxidation → cell membrane destruction
Step 3 - Gut-liver axis disruption:
- Alcohol damages the gut lining → bacteria and their products (especially lipopolysaccharide, LPS) leak from gut into portal blood → reach the liver → activate Kupffer cells (the liver's resident macrophages)
- Activated Kupffer cells release inflammatory cytokines: TNF-α, IL-1β, IL-6, IL-8
- These cytokines cause further hepatocyte death and activate stellate cells
Step 4 - Stellate cell activation → Fibrosis:
- Hepatic stellate cells (Ito cells) normally store vitamin A in a quiescent state
- When activated by inflammation signals, they transform into myofibroblasts
- Myofibroblasts produce massive amounts of Type I collagen → laid down as scar tissue = fibrosis
- Continued injury → fibrosis → cirrhosis
Why AST > ALT in Alcoholic Hepatitis:
- Alcohol depletes pyridoxal phosphate (Vitamin B6) - a cofactor that ALT needs more than AST
- So ALT production is disproportionately reduced
- Alcohol-damaged mitochondria release mitochondrial AST, raising total AST further
- Classical finding: AST:ALT ratio >2:1 (rarely exceeds 300 U/L total)
Why Does Acetaminophen (Paracetamol) Cause Hepatitis?
Normally, paracetamol is metabolized safely via glucuronidation and sulphation. A small fraction goes through CYP2E1 to form a toxic metabolite called NAPQI (N-acetyl-p-benzoquinone imine). This NAPQI is immediately detoxified by glutathione in normal doses.
In overdose: So much NAPQI is formed that glutathione stores are depleted. Free NAPQI then binds covalently to liver cell proteins → disrupts mitochondrial function → Zone 3 (centrilobular) necrosis (this zone has the highest CYP2E1 activity and lowest glutathione reserves).
This is why N-acetylcysteine (NAC) is the antidote - it replenishes glutathione stores.
SECTION 4: PATHOPHYSIOLOGY (Step-by-Step)
The Universal Final Common Pathway
Regardless of cause, hepatitis follows the same general pathway of injury:
Trigger (virus/toxin/immune attack)
↓
Hepatocyte damage (necrosis/apoptosis)
↓
Intracellular enzymes (ALT, AST) leak into blood → elevated LFTs
↓
Kupffer cell activation → cytokine storm → inflammation
↓
Bilirubin processing disrupted → jaundice
↓
Synthetic function impaired → low albumin, prolonged PT
↓
Ammonia accumulation → encephalopathy (if severe)
↓
Stellate cell activation → collagen deposition → fibrosis
↓
(If chronic/repeated injury) → Cirrhosis → Portal hypertension → Complications
Pathophysiology of HBV in Detail
Phase 1 - Viral Entry
- HBV circulates in blood
- HBV uses its surface proteins (Pre-S1) to bind to the NTCP receptor (sodium-taurocholate cotransporting polypeptide) on the hepatocyte surface
- This is the "lock-and-key" entry point; bile acid transporter hijacked by the virus
- Virus enters and releases its DNA into the nucleus
Phase 2 - Establishment of cccDNA (The Master Template)
- The partially double-stranded HBV DNA is repaired inside the nucleus to form cccDNA
- cccDNA is like a permanent hard drive inside the cell - it cannot be eradicated by current antivirals
- This is why HBV cannot be fully cured (only suppressed) - cccDNA persists even when blood viral load is undetectable
Phase 3 - Viral Gene Expression
- cccDNA transcribes several RNA products:
- Pregenomic RNA (pgRNA) → packaged and reverse-transcribed into new HBV DNA
- mRNAs → translated into HBsAg, HBcAg, HBeAg, and X protein
- HBeAg is secreted into blood as a "decoy" - it may suppress immune response by inducing T-cell tolerance
Phase 4 - Immune Recognition (Key Step)
- The immune system is NOT fighting the virus directly - it is fighting the infected hepatocytes
- CD8+ cytotoxic T lymphocytes (CTLs) circulate and scan cells
- Infected hepatocytes display viral peptides on their MHC class I molecules
- CTLs recognize these peptides → activate → kill the infected hepatocytes (via perforin-granzyme and Fas-FasL pathways)
- This killing = liver cell necrosis = the rise in ALT/AST
- The more vigorous the CTL response, the worse the hepatitis BUT the better the chance of clearing the virus
Phase 5 - Fibrosis Development (Chronic HBV)
- In chronic infection, repeated rounds of hepatocyte death → healing → scar
- Dead hepatocytes release TGF-β, PDGF
- These cytokines activate hepatic stellate cells (HSCs)
- HSCs transform from fat-storing cells into collagen-producing myofibroblasts
- Type I and III collagen is deposited in the Space of Disse (between hepatocytes and sinusoids)
- This disrupts normal liver architecture → bands of scar tissue form between portal tracts = fibrosis
- Eventually the entire architecture is replaced by regenerative nodules surrounded by fibrosis = cirrhosis
Phase 6 - HCC (Hepatocellular Carcinoma) Risk
- Two mechanisms:
- Indirect: Repeated hepatocyte death → regeneration → proliferating cells accumulate mutations → cancer
- Direct: HBV's X protein (HBx) is a transcriptional transactivator - it activates proto-oncogenes, inhibits p53 tumour suppressor, and promotes cell survival signalling → can directly drive malignant transformation even without cirrhosis
Pathophysiology of HCV in Detail
Why HCV Is Harder to Clear Than HAV
- HCV's RNA-dependent RNA polymerase has no proofreading ability → generates ~10^12 viral particles per day with very high mutation rate
- This creates a swarm of slightly different variants (quasispecies) → immune system cannot keep up
- HCV also specifically blocks innate immune signalling:
- NS3/4A protease cleaves TRIF and MAVS (key adaptor proteins in interferon signalling pathways) → prevents interferon production
- Less interferon = less antiviral defence = virus survives
Liver Damage Mechanism in HCV
- HCV is partially directly cytopathic (more so than HBV) AND causes immune-mediated damage
- The steatosis seen in HCV (especially genotype 3) is caused by HCV core protein disrupting lipid metabolism in hepatocytes
- CD4+ and CD8+ T-cells attack infected hepatocytes → necro-inflammation → fibrosis
- Fibrosis in HCV typically progresses through 5 stages (METAVIR: F0-F4):
- F0: No fibrosis
- F1: Portal fibrosis without septa
- F2: Portal fibrosis with rare septa
- F3: Numerous septa without cirrhosis
- F4: Cirrhosis
- Average time from infection to cirrhosis: 20-30 years (faster with alcohol, HIV co-infection, older age at infection)
Pathophysiology of Autoimmune Hepatitis
- The immune system incorrectly recognizes liver self-antigens as foreign
- Molecular mimicry: Viral or environmental triggers cause the immune system to produce antibodies and T-cells that cross-react with hepatocyte proteins
- Type 1 AIH: Antibodies against smooth muscle (anti-SMA) and nuclear antigens (ANA) - attack hepatocyte cytoskeleton and nuclear proteins
- Type 2 AIH: Anti-LKM1 antibodies - target CYP2D6 enzyme inside hepatocytes
- CD4+ T-helper cells and CD8+ CTLs infiltrate portal tracts → interface hepatitis (inflammation at the junction of portal tract and liver parenchyma) = the histological hallmark
- Because the attack is immune-mediated, it responds to immunosuppression (steroids, azathioprine)
The Final Common Pathway: How Cirrhosis Causes All Its Complications
Once cirrhosis develops, these complications arise mechanically:
Portal Hypertension
- Normal portal vein pressure: 5-10 mmHg
- Fibrosis and nodule formation distort the intrahepatic vasculature → increased resistance to blood flow
- Portal vein pressure rises to >12 mmHg = portal hypertension
- Blood seeks alternative routes (collaterals) to drain into systemic circulation
Oesophageal Varices
- Portal blood diverts through the left gastric vein → oesophageal submucosal veins → azygos vein → systemic
- These thin-walled veins become dilated and tortuous = varices
- They are superficial and fragile → rupture → massive haematemesis → 10-15% mortality per episode
Ascites
- Portal hypertension → increased hydrostatic pressure in splanchnic capillaries → fluid leaks into peritoneal cavity
- Low albumin (synthetic failure) → reduced oncotic pressure → fluid stays out
- Portal hypertension triggers splanchnic vasodilation → renin-angiotensin-aldosterone system activates → sodium and water retention → fluid accumulates
Hepatic Encephalopathy
- Normal liver converts toxic ammonia (from gut bacterial metabolism of amino acids + nitrogenous waste) → urea → excreted in urine
- In cirrhosis: liver cells fail + blood shunts bypass the liver through portosystemic collaterals
- Ammonia accumulates in blood → crosses blood-brain barrier → converted to glutamine in astrocytes → astrocyte swelling → cerebral oedema
- Ammonia also disrupts neuronal inhibitory/excitatory balance (increases GABAergic tone) → altered consciousness
- Precipitants: GI bleeding (large protein load from blood in gut), infection, dehydration, hypokalemia (raises renal ammonia production), constipation
Hepatorenal Syndrome (HRS)
- Splanchnic vasodilation (from portal hypertension) → blood pools in gut vessels → effective circulating volume falls
- Kidneys sense reduced perfusion → vasoconstriction of renal arterioles (via angiotensin II, vasopressin, sympathetic activation)
- Renal blood flow drops → acute kidney injury without intrinsic kidney disease
- The kidneys are structurally normal (proven by the fact that if transplanted into a healthy person, they work fine)
- Type 1 HRS: Rapid (creatinine doubles to >226 μmol/L in <2 weeks) - very high mortality
- Type 2 HRS: Slower, associated with refractory ascites
Hepatocellular Carcinoma (HCC)
- Repeated cell death → regeneration → replicating cells accumulate DNA mutations
- Hepatocytes in cirrhosis are in a constant cycle of death and proliferation
- Mutations in tumour suppressor genes (p53, Rb) and proto-oncogenes (CTNNB1, TERT) accumulate
- HBV additionally has HBx protein that directly promotes oncogenesis
- HCC can arise even without cirrhosis in HBV (unique among liver diseases)
SECTION 5: CLINICAL FEATURES - WHY EACH SYMPTOM OCCURS
| Symptom/Sign | Mechanism (Why It Happens) |
|---|---|
| Fatigue/malaise | Cytokine release (IL-1, TNF-α, IL-6) during immune activation has systemic effects on the brain - the same molecules that cause fever also cause fatigue and malaise ("sickness behaviour") |
| Anorexia/nausea | Same cytokines act on the hypothalamus; also bile backing up into the stomach; altered gut motility |
| RUQ pain | Rapid hepatocyte swelling → liver capsule (Glisson's capsule) is stretched - the capsule has pain-sensitive nerve fibres; the liver parenchyma itself has no pain fibres |
| Fever | Cytokines (especially IL-1β and IL-6) reach the hypothalamus → reset the thermoregulatory set-point upward → fever |
| Arthralgia/myalgia | Immune complex deposition in joints and muscles (especially HBV - immune complexes of HBsAg + anti-HBs + complement); also direct cytokine effects on muscles |
| Jaundice | Hepatocytes are damaged → cannot conjugate bilirubin OR conjugated bilirubin cannot be excreted into bile canaliculi → conjugated bilirubin accumulates in blood → deposits in skin and sclera = yellow |
| Dark urine | Conjugated bilirubin is water-soluble → spills into kidneys → appears in urine → tea/cola-coloured urine. Only conjugated bilirubin appears in urine (unconjugated is bound to albumin and cannot be filtered) |
| Pale stools | Bilirubin normally gives stool its brown colour (stercobilin). When bilirubin cannot reach the gut (because excretion is blocked), stools lose their colour = clay-coloured/pale stools |
| Pruritus (itch) | Bile salts accumulate in the skin (cannot be excreted properly). Bile salts activate itch-specific nerve fibres (pruritoceptors) in the dermis. Also: lysophosphatidic acid (LPA) is produced and activates itch fibres |
| Hepatomegaly | Inflammation → oedema → liver swells. Hepatocytes are being killed and replaced; Kupffer cells are activated; inflammatory infiltrate fills portal tracts → liver enlarges |
| Splenomegaly | Portal hypertension → backed-up blood in splenic vein → spleen enlarges. Also, in viral illness, the spleen is actively fighting infection (B-cell and T-cell expansion). Hypersplenism results in thrombocytopenia (platelets trapped and destroyed in enlarged spleen) |
| Asterixis | Ammonia disrupts neuronal transmission, especially motor control areas. The abnormal electrical activity in the brain causes brief lapses in postural muscle tone = the characteristic flapping tremor when wrists are extended |
| Ascites | Combination of portal hypertension (fluid driven out of capillaries) + hypoalbuminaemia (reduced oncotic pressure to hold fluid in vessels) + sodium retention (RAAS activation) |
| Spider angiomata / Palmar erythema | Liver normally metabolises oestrogen. In cirrhosis, oestrogen accumulates → vasodilation → spider naevi (central arteriole with radiating vessels) on upper body and chest; diffuse erythema of palms |
| Coagulopathy / easy bruising | Liver synthesises all clotting factors (I, II, V, VII, IX, X, XI) except Factor VIII. In severe hepatitis, factor production falls → PT and APTT prolong → bruising, bleeding tendency |
| Gynaecomastia | Excess oestrogen (not metabolised) stimulates breast tissue growth in men |
SECTION 6: INVESTIGATIONS - FULLY EXPLAINED
Why We Do Each Test and What Each Result Tells Us
ALT (Alanine Aminotransferase)
- Location: Found in high concentrations in hepatocyte cytoplasm
- Why it rises: When hepatocytes die (necrosis or apoptosis), this enzyme leaks out into the blood. Think of it like breaking open a bag of flour - the contents spill everywhere.
- Why ALT > AST in viral hepatitis: ALT has a longer half-life than AST (47 hours vs 17 hours). Also, in viral hepatitis, cytoplasmic release of ALT predominates. ALT is more specific to the liver (AST also exists in heart, muscle, red blood cells).
- Expected level: 8-50× upper limit of normal (ULN) in acute viral hepatitis. An ALT >300 U/L has >90% sensitivity for acute hepatitis.
- Why it matters: Best real-time marker of hepatocyte injury. Used to monitor treatment response - as hepatocytes recover, ALT falls back to normal.
AST (Aspartate Aminotransferase)
- Location: Hepatocyte cytoplasm AND mitochondria; also in heart, skeletal muscle, red blood cells
- AST:ALT ratio > 2:1 = strongly suggests alcoholic hepatitis because:
- Mitochondrial AST is released specifically by alcohol-damaged mitochondria
- Alcohol depletes Vitamin B6, which ALT needs more than AST for its enzymatic function
- In shock liver (ischaemic hepatitis): Both AST and ALT can shoot up to 50-100× ULN but then fall rapidly within 48-72 hours as hypoperfusion resolves
ALP (Alkaline Phosphatase)
- Location: Canalicular surface of hepatocytes AND bile duct epithelium; also in bone, placenta, intestine
- Why it rises: ALP is embedded in the bile canaliculi. When bile flow is obstructed (inside or outside the liver), bile pressure rises → ALP is "washed back" into blood. Also, bile acids and bilirubin stimulate ALP synthesis.
- Why it is only mildly elevated in hepatitis (<3× ULN in 90%): The hepatocyte injury in hepatitis is primarily in the liver parenchyma, not the biliary system. Severe cholestatic obstruction (gallstone, tumour) causes ALP >3× ULN.
- Differentiating hepatic vs bone source of elevated ALP: Check GGT - if GGT is also high, the source is hepatic; if GGT is normal, suspect bone (e.g., Paget's disease)
GGT (Gamma-Glutamyl Transferase)
- Location: Liver, kidneys, pancreas - most sensitive in the liver
- Why it rises with alcohol: Alcohol is a potent inducer of CYP enzymes and also of GGT synthesis. GGT rises even with moderate, regular alcohol use.
- Why it's clinically important: Disproportionately high GGT relative to other LFTs strongly suggests alcohol as the cause even if the patient denies drinking
Serum Bilirubin (Total, Direct/Conjugated, Indirect/Unconjugated)
The Bilirubin Journey - Understanding the Whole Picture:
Red blood cells break down (reticuloendothelial system, especially spleen)
↓
Haemoglobin → Haem + Globin
↓ (Kupffer cells in liver; macrophages in spleen)
Haem → Biliverdin → Unconjugated bilirubin (lipid-soluble, bound to albumin in blood)
↓ (enters hepatocytes)
Conjugated to glucuronic acid by UGT1A1 enzyme → Conjugated bilirubin (water-soluble)
↓
Excreted into bile canaliculi → Bile ducts → Small intestine
↓
Intestinal bacteria convert to urobilinogen → Stercobilin (brown colour of stool)
↓ (small amount reabsorbed)
Urobilinogen → Blood → Kidneys → Urine (gives urine its pale yellow colour)
- In hepatitis: The conjugating machinery is intact BUT the excretion of conjugated bilirubin into bile is impaired (injured hepatocytes cannot pump bilirubin into canaliculi). Conjugated bilirubin refluxes back into the blood.
- Why dark urine: Conjugated bilirubin is water-soluble → kidneys filter it → dark urine
- Why pale stools: Less bilirubin reaching the gut → less stercobilin → pale stools
- Why jaundice appears at bilirubin >35-50 μmol/L: This is the threshold at which tissues (especially sclera, which has high elastin that binds bilirubin well) become visibly yellow
Albumin
- Produced exclusively by the liver (the main protein synthesiser in the body)
- Half-life ~21 days - very long. Albumin is like a long-running project; it takes time to fall.
- Why it is NORMAL in acute hepatitis: Even with significant acute hepatitis, the liver still has enough residual function to maintain albumin levels for weeks. Albumin falls only when >80% of functional liver tissue is destroyed.
- Why it falls in chronic hepatitis/cirrhosis: Prolonged loss of hepatocytes → sustained reduction in albumin synthesis
- Clinical consequence of low albumin: Oncotic pressure drops → fluid leaks out of vessels → oedema and ascites
PT/INR (Prothrombin Time)
- The liver makes almost all clotting factors: I (fibrinogen), II (prothrombin), V, VII, IX, X, XI, XIII
- Factor VII has the shortest half-life (~6 hours) → PT/INR is the EARLIEST and MOST SENSITIVE marker of acute liver synthetic failure
- In acute liver failure, PT/INR is the single best prognostic marker - if it continues to rise despite treatment, it indicates the liver is losing the battle
- PT prolongation means the time it takes for blood to clot is longer → bleeding risk
Serum Ammonia
- Ammonia is produced in the gut (bacterial protein metabolism) and enters the portal blood
- Normal liver: Converts ammonia → urea via Ornithine Cycle (urea cycle) → excreted in urine
- Cirrhotic liver: Reduced urea cycle activity + portosystemic shunting (blood bypasses liver) → ammonia accumulates
- In encephalopathy: Ammonia enters the brain → astrocytes (the brain's housekeeping cells) take it up and use it to convert glutamate → glutamine (a detoxification mechanism) → but glutamine accumulation causes astrocyte swelling → cerebral oedema → encephalopathy
Serological Tests - What They Mean and WHY
Hepatitis A Serology
IgM Anti-HAV:
- IgM is the FIRST antibody the immune system makes against any new infection (appears within 1-2 weeks of infection)
- Positive IgM = the body just encountered HAV = ACUTE infection
- IgM disappears after 3-6 months
IgG Anti-HAV:
- IgG comes later (weeks after IgM) and persists for life
- Positive IgG = past infection OR successful vaccination = IMMUNITY
- You cannot tell the difference between natural immunity and vaccine immunity from IgG alone
Hepatitis B Serology - The Full Map
HBsAg (Hepatitis B Surface Antigen):
- This is a protein on the surface of HBV. When the virus is replicating, it makes massive excess amounts of HBsAg particles (even more than actual virus).
- Positive HBsAg = virus is present = active infection (acute or chronic)
- Persistent >6 months = chronic HBV infection
- HBsAg alone cannot distinguish acute from chronic - you need IgM anti-HBc for that
IgM Anti-HBcAg (IgM Antibody to Core Antigen):
- HBcAg (core antigen) is NEVER found free in blood (it is inside the viral particle). But the immune response to it is detectable.
- IgM anti-HBc appears early in acute infection and persists for ~6 months
- This is the definitive marker of ACUTE HBV infection
- It also becomes positive in reactivation of chronic HBV (important to know)
Anti-HBc Total (IgG):
- Persists for life after any HBV infection, whether resolved or chronic
- If this is positive but HBsAg is negative and anti-HBsAg is positive → past resolved infection with immunity
- If this is positive and HBsAg is positive → chronic infection ongoing
Anti-HBsAg:
- Appears after HBsAg clears (recovery) or after vaccination
- This is the protective antibody - it neutralises the virus
- Level >10 mIU/mL = protected
- Only anti-HBsAg is positive (with negative anti-HBc) = vaccinated person (not natural infection, as vaccination only generates surface antigen antibodies)
HBeAg:
- A secreted form of the core protein, released by highly replicating hepatocytes
- Presence = high viral replication = high infectivity
- It also acts as an immunological decoy - tricks the immune system into tolerating the virus
Anti-HBeAg:
- Appears when HBeAg is cleared (seroconversion)
- Marks transition from active replication to lower replication
- However, HBeAg-negative chronic HBV still exists (precore mutant virus - does not make HBeAg but still replicates dangerously)
HBV DNA (Quantitative PCR):
- Directly measures viral load in blood
- Gold standard for monitoring antiviral therapy
- Used to determine whether treatment is needed and whether it is working
Hepatitis C Serology
Anti-HCV (ELISA):
- Detects antibodies to HCV proteins
- Window period: Antibodies may not appear until 8-12 weeks after infection → early acute HCV can be missed
- Positive = EXPOSED to HCV at some point (does NOT mean currently infected - could be past cleared infection)
- Does NOT distinguish acute from chronic from past cleared infection
HCV RNA (PCR):
- Detects actual viral genetic material in blood
- Becomes detectable within 1-2 weeks of infection (much earlier than antibody)
- Used to: confirm active infection, determine viral load (predicts treatment success), and monitor treatment
- Undetectable HCV RNA at 12 weeks post-treatment = SVR (Sustained Virologic Response) = CURE
HCV Genotype (1-6):
- Different strains of HCV (like different subspecies)
- Genotype determines which DAA regimen to use and how long to treat
- Genotype 3: More fibrosis, more steatosis, harder to treat historically (though modern pan-genotypic regimens work well)
Liver Biopsy - Why It Remains Important
Even with good serology and imaging, the biopsy tells us what serological tests cannot:
- How inflamed is the liver right now? (Grade of necroinflammation: G0-G4)
- How much scar tissue is there? (Stage of fibrosis: F0-F4 or Ishak 0-6)
- What does the cause look like histologically? (AIH has interface hepatitis; alcoholic hepatitis has Mallory-Denk bodies; NASH has steatohepatitis)
- Are there features of malignant transformation?
The grade and stage together determine prognosis and whether treatment is urgent.
Imaging
Ultrasound - The First-Line Tool
- Non-invasive, cheap, no radiation
- In acute hepatitis: Liver enlarges → decreased echogenicity (looks darker) compared to the kidney. Portal tracts become echogenic (look bright) against the dark background = "Starry sky" pattern (like stars in a dark sky). This is classic but not pathognomonic.
- In chronic hepatitis: Echogenicity increases (liver looks coarser and brighter) as fibrosis replaces normal tissue; the portal vein radicles become less visible as they are surrounded by fibrosis
- Detects cirrhosis (nodular surface, right lobe atrophy, caudate/left lobe hypertrophy), ascites, splenomegaly, dilated portal vein
MRI - Most Sensitive for Acute Hepatitis
- Gadolinium-enhanced MRI during the late arterial phase (18-22 seconds after injection) shows periportal enhancement in acute hepatitis
- T2-weighted images show periportal oedema as increased signal
- More expensive than ultrasound but more sensitive for subtle disease
Fibroscan (Transient Elastography)
- Sends a vibration wave into the liver; measures how stiff the liver is
- Stiff liver = more fibrosis
- Provides a liver stiffness measurement (kPa) that correlates with METAVIR fibrosis stage
- Has replaced liver biopsy for staging fibrosis in many chronic hepatitis patients
SECTION 7: COMPLICATIONS - FULLY EXPLAINED
How Each Complication Develops
Variceal Bleeding
- When portal pressure exceeds 12 mmHg, varices form
- When portal pressure exceeds 20 mmHg, risk of rupture is very high
- The risk of bleeding from a varix is directly proportional to its size and the presence of red wale signs (red streaks on varix surface indicating wall thinning)
- When a varix ruptures: massive haemorrhage into the oesophageal or gastric lumen → haematemesis (vomiting blood) or melaena (black tarry stools from digested blood)
- Bleeding itself raises ammonia (blood in the gut is a protein load → bacteria metabolise it → ammonia rises) → can trigger encephalopathy
Spontaneous Bacterial Peritonitis (SBP)
- In cirrhosis, the immune defence of the peritoneal fluid is impaired (low complement, low opsonic activity)
- Gut bacteria translocate across the damaged intestinal wall into lymphatics → portal blood → ascitic fluid
- Without normal immune defences, bacteria multiply in ascitic fluid
- Diagnosis: Ascitic fluid neutrophil count >250 cells/mm³ (even before culture results)
- Treatment: Third-generation cephalosporin (ceftriaxone) + albumin infusion (prevents HRS)
Hepatocellular Carcinoma
- Once cirrhosis is established, HCC risk = 1-8% per year depending on aetiology (HBV highest)
- HBV can cause HCC even WITHOUT cirrhosis (unique - due to HBx protein)
- Surveillance: 6-monthly liver ultrasound + serum AFP (alpha-fetoprotein) in all cirrhotic patients and HBV carriers
- AFP elevation suggests HCC but is not diagnostic alone (can be elevated in regenerative nodules)
- Diagnosis confirmed by contrast CT/MRI showing arterial enhancement + washout of a liver lesion (no biopsy needed if imaging is typical)
SECTION 8: PREVENTION - WHY EACH MEASURE WORKS
Hepatitis A Vaccine
- Formalin-inactivated HAV is injected → your immune system sees the dead virus → produces IgG anti-HAV antibodies and memory B cells
- If you later encounter live HAV, your memory B cells rapidly produce antibodies before the virus can establish infection → sterilising immunity
- Two doses give >95% seroprotection for at least 25 years (likely lifetime)
Hepatitis B Vaccine
- Contains recombinant HBsAg (produced by genetically modified yeast - no live virus)
- Immune system makes anti-HBsAg antibodies
- Anti-HBs >10 mIU/mL = protective threshold
- Three-dose schedule (0, 1, 6 months) builds robust, long-lasting immunity
- Universal infant vaccination is the single most impactful intervention against HBV worldwide - it reduces HBV transmission by 95% in immunised populations
- Immunosuppressed patients (dialysis, chemotherapy, HIV) respond poorly → may need higher doses or additional doses
Post-Exposure Prophylaxis for HBV
HBIG (Hepatitis B Immunoglobulin):
- This is pre-formed anti-HBsAg antibody from pooled human plasma of vaccinated donors
- It provides immediate passive immunity - neutralises circulating HBV before it can establish infection
- Must be given within 96 hours (ideally <24 hours) for maximum effect
- Given together with vaccine to bridge the gap until the vaccine-induced active immunity develops
Why There Is No HCV Vaccine
- HCV mutates rapidly (quasispecies) → no stable surface antigen to target
- Past infection does NOT confer immunity to re-infection (unlike HAV and HBV)
- Any vaccine-induced antibodies are quickly outpaced by new viral variants
- Research is ongoing but progress is very slow
Harm Reduction for HCV
- Needle exchange programs: Remove the contaminated needle as the transmission vector
- Opioid substitution therapy (methadone, buprenorphine): Reduces injecting frequency → reduces HCV transmission
- These are population-level interventions that have demonstrably reduced new HCV infections in multiple countries
SECTION 9: MANAGEMENT - HOW EVERY TREATMENT WORKS
Non-Pharmacological Management
Rest
- During acute hepatitis, liver blood flow and metabolic demands matter
- Physical activity diverts blood to muscles away from the liver
- Rest maximises hepatic perfusion and provides optimal conditions for hepatocyte regeneration
- However, prolonged bed rest is not necessary - activity as tolerated is the modern approach
Diet
- High carbohydrate, moderate protein, low fat: The injured liver struggles with fat metabolism; carbohydrates provide easy energy. Protein is needed for cell repair but excess raises ammonia in encephalopathy.
- Avoid alcohol completely: Even small amounts cause further mitochondrial damage in an already inflamed liver
- No paracetamol: Depletes glutathione reserves in an already stressed liver
- Thiamine supplementation in alcoholic hepatitis (prevents Wernicke's encephalopathy - a brain complication of B1 deficiency)
Managing Ascites
- Salt restriction (<88 mmol sodium/day): Reduces osmotic drive for fluid retention
- Spironolactone (first-line diuretic): Blocks aldosterone → kidney stops retaining sodium/water → fluid excreted
- Furosemide (loop diuretic): Added if spironolactone alone insufficient
- Therapeutic paracentesis: For large-volume ascites (>5L): drain fluid with a needle. Must give albumin (8g per litre removed) to prevent circulatory dysfunction and hepatorenal syndrome
Managing Variceal Bleeding
- Terlipressin: A vasopressin analogue → causes splanchnic vasoconstriction → reduces portal pressure → slows variceal bleeding. First given in the ambulance/ER.
- Endoscopic variceal banding (EVL): A rubber band is placed around the varix → strangulates it → varix thromboses and falls off
- Propranolol / Carvedilol: Non-selective beta-blockers for PRIMARY prophylaxis → reduce cardiac output AND cause splanchnic vasoconstriction → lower portal pressure
Pharmacological Management
How Antivirals Work - Step by Step
Tenofovir (TDF/TAF) - For Chronic HBV
- Class: Nucleotide analogue (mimics adenosine monophosphate)
- Mechanism: Tenofovir is taken up by hepatocytes → phosphorylated to its active triphosphate form → competes with the natural substrate (dATP) for binding to HBV's reverse transcriptase (the enzyme that copies viral RNA into DNA)
- Once incorporated into the growing HBV DNA chain, it acts as a chain terminator (lacks the 3'-OH group needed for the next nucleotide to attach) → viral DNA synthesis stops → no new virions can be made
- Why it doesn't cure HBV: Tenofovir does NOT affect the cccDNA already established in the nucleus - it only prevents production of NEW viral copies
- TAF vs TDF: TAF is a prodrug that delivers tenofovir more efficiently to liver cells (a "targeted delivery" form), so a much lower dose is needed, with less systemic exposure → less kidney toxicity, less bone density loss
Entecavir - For Chronic HBV
- Class: Guanosine nucleoside analogue
- Mechanism: Competitively inhibits three steps of HBV reverse transcriptase:
- Priming of pgRNA
- Reverse transcription (RNA → DNA)
- Synthesis of the positive strand DNA
- Very high barrier to resistance - requires three simultaneous mutations for HBV to escape → almost no resistance with long-term use (unlike older agents like lamivudine)
Pegylated Interferon-alfa-2a - For Chronic HBV/HCV
- Mechanism: Not directly antiviral - it works by boosting the immune system's natural defences
- Interferon-α binds to interferon receptors on liver cells → activates the JAK-STAT signalling pathway → nucleus produces interferon-stimulated genes (ISGs)
- ISGs create an antiviral state: degrade viral RNA, block protein synthesis, inhibit viral budding
- Also activates NK cells and enhances MHC class I expression (helps CTLs find and kill infected hepatocytes)
- "Pegylated" = attached to polyethylene glycol molecule → slows its removal from blood → allows once-weekly dosing instead of three times daily
- Advantages: Finite course (48 weeks); can achieve functional cure (HBsAg loss) in a subset; no resistance
- Disadvantages: Many side effects (flu symptoms, depression, hair loss, cytopenia, thyroid dysfunction) because you are essentially creating a prolonged artificial "infection-fighting" state
Sofosbuvir - For Chronic HCV
- Class: NS5B RNA-dependent RNA polymerase inhibitor (nucleotide analogue)
- Mechanism: HCV uses its own RNA polymerase (NS5B) to copy its genome. Sofosbuvir mimics uridine → gets incorporated into the growing HCV RNA chain → chain termination → no new HCV RNA copies
- HCV has NO proofreading on its RNA polymerase → theoretically lots of resistance. BUT sofosbuvir resistance requires very specific mutations in the polymerase that severely cripple viral fitness → very rare
NS5A Inhibitors (Ledipasvir, Velpatasvir, Pibrentasvir)
- Target: NS5A protein - a non-structural HCV protein with no known enzymatic function but essential for viral replication, assembly, and secretion
- NS5A inhibitors bind to this protein → disrupt the membranous web (the intracellular compartment where HCV replicates) → virus cannot assemble or be secreted
- Ultra-potent: Can reduce HCV viral load by 4-5 log₁₀ within days
- Combined with sofosbuvir (hit two different targets simultaneously) → synergistic effect + prevents resistance from emerging
Glecaprevir - NS3/4A Protease Inhibitor
- HCV uses a protease (NS3/4A) to cleave a large viral polyprotein into individual functional proteins
- Without cleavage, the proteins remain fused and non-functional
- Glecaprevir inhibits this protease → viral proteins cannot be processed → no functional virions
Why DAA Combinations Work So Well (>95% SVR)
- Each drug hits a different step in HCV's lifecycle
- For resistance to emerge, HCV would need to simultaneously develop resistant mutations to all three targets
- The probability of three simultaneous resistance mutations is essentially zero at any given replication cycle
- This is the same principle as combination HIV therapy (HAART)
Steroids for Autoimmune Hepatitis - Mechanism
- Prednisolone suppresses immune activation by:
- Entering immune cells → binds glucocorticoid receptor → enters nucleus → suppresses NF-κB (the master switch for inflammatory gene transcription)
- Reduces production of IL-1, IL-2, IL-6, TNF-α, and interferon-γ
- Causes lymphocyte redistribution (lymphocytes exit blood → less immune attack on liver)
- Azathioprine (maintenance therapy):
- Converted to 6-mercaptopurine → inhibits purine synthesis → lymphocytes CANNOT proliferate without purines (they rely on de novo synthesis unlike other cells)
- Specifically reduces CD4+ T-helper cell activation → less immune attack
- Used at lower doses for maintenance (fewer side effects than prolonged steroids)
N-Acetylcysteine (NAC) for Acetaminophen Toxicity
- Mechanism: NAC is a glutathione precursor. It donates cysteine for glutathione synthesis.
- Replenishes hepatic glutathione stores → newly produced glutathione neutralises NAPQI before it can bind to proteins
- Must be given early (within 8-10 hours ideally for maximum effect; still beneficial up to 24 hours)
- Also used in non-acetaminophen acute liver failure: appears to improve outcomes, possibly by reducing oxidative stress and improving mitochondrial function
Lactulose for Hepatic Encephalopathy
- Mechanism (dual):
- Osmotic laxative: Non-absorbable disaccharide → draws water into the colon → diarrhoea → rapid clearance of ammonia-producing bacteria and nitrogenous waste from the gut
- Acidification of colon: Gut bacteria convert lactulose to lactic and acetic acid → lowers colonic pH → ammonia (NH₃) is trapped as ammonium (NH₄⁺) which is charged and cannot be absorbed → excreted in stool
- Target: 2-3 soft stools per day
Rifaximin
- Non-absorbable antibiotic (stays in gut)
- Kills ammonia-producing gut bacteria (specifically targets bacterial RNA polymerase)
- Reduces gut ammonia production → reduces hepatic encephalopathy recurrence
- Used as add-on to lactulose for prevention of recurrent HE
SECTION 10: DIAGNOSTIC CRITERIA - HOW TO APPLY THEM
Acute Hepatitis - No Formal Criteria; Clinical + Lab Diagnosis
You diagnose acute hepatitis when:
- Compatible symptoms (prodrome + jaundice)
- ALT/AST elevated 8-50× (or >300 U/L)
- ALP mildly elevated (<3× ULN)
- Specific serology confirms the agent
Key rule: If serology is all negative, you MUST search for drug/toxic cause - DILI is the most common missed diagnosis.
Chronic Hepatitis - Definition-Based
- Same abnormalities persisting beyond 6 months
- Graded (inflammation) and staged (fibrosis) by biopsy or non-invasive tools
Autoimmune Hepatitis Simplified Scoring (IAIHG)
| Variable | 1 Point | 2 Points |
|---|---|---|
| Autoantibodies (ANA/SMA) | ≥ 1:40 | ≥ 1:80 |
| Or Anti-LKM1 | - | ≥ 1:40 |
| Or Anti-SLA | - | Any positive |
| IgG | > ULN | > 1.1 × ULN |
| Histology | Compatible | Typical |
| Absence of viral hepatitis | - | 2 points |
- Score 6 = Probable AIH; Score ≥7 = Definite AIH
- The scoring makes it systematic - you cannot diagnose AIH from just one positive antibody
Maddrey Discriminant Function (MDF) for Alcoholic Hepatitis
MDF = 4.6 × (PT_patient - PT_control in seconds) + Serum Bilirubin (mg/dL)
- MDF < 32: Mild-moderate disease; mortality ~15%; supportive care only
- MDF ≥ 32: Severe disease; 1-month mortality ~35%; treat with prednisolone 40 mg/day
- If no improvement after 7 days of steroids (Lille score calculation) → steroids are not working → stop them
SECTION 11: DIFFERENTIAL DIAGNOSIS - DETAILED REASONING
When a patient presents with jaundice and elevated LFTs, this is your systematic approach:
Step 1: Is This Hepatocellular or Cholestatic?
| Pattern | ALT/AST | ALP | Suggests |
|---|---|---|---|
| Hepatocellular | Very high (>5× ULN) | Mild (<3×) | Hepatitis (viral, toxic, alcoholic, autoimmune, ischaemic) |
| Cholestatic | Mild (<3×) | Very high (>3-10×) | Bile duct obstruction (gallstone, cholangiocarcinoma, PSC) |
| Mixed | Both elevated | Both elevated | Drug-induced, primary biliary cholangitis |
Step 2: Work Through Each Differential
1. Viral Hepatitis A
- FOR: Ate raw shellfish/contaminated food recently; travelled to endemic area (Africa, South Asia); fecal-oral exposure; IgM anti-HAV positive; prodrome + jaundice
- AGAINST: IV drug use or sexual exposure as sole risk factor; HBsAg or anti-HCV positive; chronic course; jaundice recurring
2. Viral Hepatitis B
- FOR: IV drug use; multiple sexual partners; tattoo/piercings; birth in endemic country; HBsAg positive; IgM anti-HBc positive (acute); serum-sickness like prodrome (urticaria + arthritis) before jaundice; can recur/become chronic
- AGAINST: Purely fecal-oral exposure; IgM anti-HAV positive; normal viral serology
3. Viral Hepatitis C
- FOR: IV drug use; blood transfusion before 1992; haemodialysis; needle-stick injury; anti-HCV positive; HCV RNA detectable; typically asymptomatic acutely; persistent mild ALT elevation
- AGAINST: No parenteral risk factor; acute jaundice (HCV is usually subclinical acutely); positive HAV/HBV serology
4. Alcoholic Hepatitis
- FOR: Heavy daily alcohol use (typically >80g/day for years); AST:ALT >2:1; neither AST nor ALT >300-500 U/L despite jaundice; fever + leucocytosis (neutrophilic); tender hepatomegaly; signs of chronic liver disease; hypoglycaemia; elevated GGT disproportionately; Mallory-Denk bodies on biopsy
- AGAINST: No alcohol history; ALT > AST; viral serology positive; ALT >1000 U/L (alcohol rarely causes this - think viral, ischaemic, or acetaminophen if >1000)
5. Drug-Induced Liver Injury (DILI)
- FOR: Started a new drug or supplement within weeks to months before symptoms; improvement after stopping the drug; eosinophilia + rash (hypersensitivity-type DILI); all viral serology negative; acetaminophen history + dose >4g/day (or lower with alcohol)
- AGAINST: No new drug exposure; viral serology positive; clinical improvement despite continuing medication
6. Autoimmune Hepatitis (AIH)
- FOR: Young woman; fluctuating transaminases over years; ANA positive, anti-SMA positive, anti-LKM1 positive; IgG elevated; other autoimmune conditions (thyroiditis, IBD); responds to steroids; interface hepatitis on biopsy; AIH score ≥6
- AGAINST: Male sex (less likely); positive viral serology explaining all abnormalities; normal immunoglobulins; no autoantibodies
7. Biliary Obstruction (Choledocholithiasis/Cholangitis)
- FOR: Colicky RUQ pain (gallstone pattern) or constant pain; ALP disproportionately elevated (>3× ULN); dilated bile ducts on ultrasound/CT; prior history of gallstones; Charcot's triad for cholangitis (fever + RUQ pain + jaundice); elevated serum amylase if pancreatitis
- AGAINST: Predominantly ALT/AST pattern; prodromal viral illness; no biliary dilation on imaging; alcohol history
8. Ischaemic Hepatitis (Shock Liver)
- FOR: Recent episode of hypotension/circulatory failure (MI, septic shock, cardiac arrest, massive bleed); extremely rapid AST/ALT rise to 50-100× ULN; rapid normalization within 48-72 hours as haemodynamics recover; bilirubin paradoxically mild given the severity of enzyme rise; LD also very high (ischaemic injury releases LD)
- AGAINST: No haemodynamic event; gradual enzyme rise; positive viral serology; persistent enzyme elevation
9. Wilson's Disease
- FOR: Young patient (<40 years, usually <30); paradoxically LOW or normal ALP (Wilson's damages hepatocytes that make ALP AND it causes haemolysis which consumes ALP substrate); Kayser-Fleischer rings in eyes (copper deposits in Descemet's membrane of cornea - seen on slit-lamp); haemolytic anaemia; neurological/psychiatric symptoms; low ceruloplasmin; raised 24-hour urinary copper; liver copper elevated on biopsy
- AGAINST: Age >40; elevated ALP; positive viral serology; no haemolysis; normal copper studies
- Clinical pearl: In Wilson's presenting as acute liver failure, ceruloplasmin may paradoxically be normal (released from dying hepatocytes temporarily raises it) - look for the ALP:bilirubin ratio (<4 strongly suggests Wilson's in ALF)
10. EBV/CMV Hepatitis
- FOR: Young adult; pharyngitis + cervical lymphadenopathy (mononucleosis features); splenomegaly; heterophile antibody (Monospot) positive; lymphocytosis with atypical lymphocytes on blood film; EBV/CMV IgM serology positive; hepatitis is mild and self-limiting; mild transaminase elevation (rarely exceeds 5× ULN)
- AGAINST: Positive HAV/HBV/HCV serology; no lymphadenopathy/pharyngitis; severe hepatitis; IV drug use
11. Non-Alcoholic Steatohepatitis (NASH/MASH)
- FOR: Metabolic syndrome (obesity, Type 2 diabetes, hypertriglyceridaemia, hypertension); AST/ALT typically <3× ULN and persistent; all viral serology negative; no significant alcohol intake; steatosis on ultrasound (bright echogenic liver); AST:ALT <1 typically; gradual, asymptomatic course
- AGAINST: Acute jaundice; significant ALT elevation >10× ULN; positive viral serology; active alcohol use (alcohol causes a similar picture but is treated as ARLD); autoantibodies positive
Key Teaching Point from Yamada's Gastroenterology: "Drug-induced liver disease should be considered in EVERY case of acute hepatitis until the etiology is firmly established. A detailed drug history is MANDATORY from every patient with acute hepatitis." The most common diagnostic error is missing DILI because no one asked about herbal supplements or over-the-counter medications.
Sources Used:
- Tietz Textbook of Laboratory Medicine, 7e - Investigations and pathogenesis
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease - Chronic hepatitis, HBV/HCV mechanisms
- Rosen's Emergency Medicine, 10e - Clinical approach, alcoholic hepatitis
- Goldman-Cecil Medicine - Antiviral drug doses and indications
- Morgan and Mikhail's Clinical Anesthesiology, 7e - Drug-induced hepatitis, acute hepatitis
- Yamada's Textbook of Gastroenterology, 7e - DDx, imaging
- Symptom to Diagnosis: An Evidence-Based Guide, 4e - AIH criteria, DDx tables
- Sherris Medical Microbiology, 8e - HDV structure and mechanism
- Jawetz Medical Microbiology, 28e - Prevention and vaccination
This is a shared conversation. Sign in to Orris to start your own chat.