Purine and pyrimidine metabolism and disorders
Reading File
Finding Sources
Finding Sources
Searching PubMed
purine pyrimidine metabolism disorders
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Finding Sources
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer:
Purine and Pyrimidine Metabolism and Disorders
1. Overview and Functions of Nucleotides
Purines and pyrimidines are the nitrogen-containing bases that form the backbone of nucleotides and nucleic acids. Beyond DNA and RNA synthesis, nucleotides serve multiple roles:
- Energy currency: ATP, GTP
- Coenzymes: NAD+, FAD, Coenzyme A
- Activated intermediates: UDP-glucose (glycogen synthesis), CDP-diacylglycerol (phospholipid synthesis)
- Second messengers: cAMP, cGMP
- Allosteric regulators of metabolic enzymes
Dietary nucleic acid contributes minimally to the nucleotide pool; pancreatic DNase and RNase digest ingested nucleic acids, but intestinal epithelial cells efficiently catabolize the products. Nucleotides must therefore be synthesized endogenously.
- Basic Medical Biochemistry, 6e, p. 1404
2. Purine Metabolism
A. De Novo Synthesis
Purines are built atom by atom on a ribose 5-phosphate scaffold. The process is complex (11 steps), energy-expensive (6 ATP per purine), and starts with PRPP (5-phosphoribosyl-1-pyrophosphate).
Precursors contributing to the purine ring:
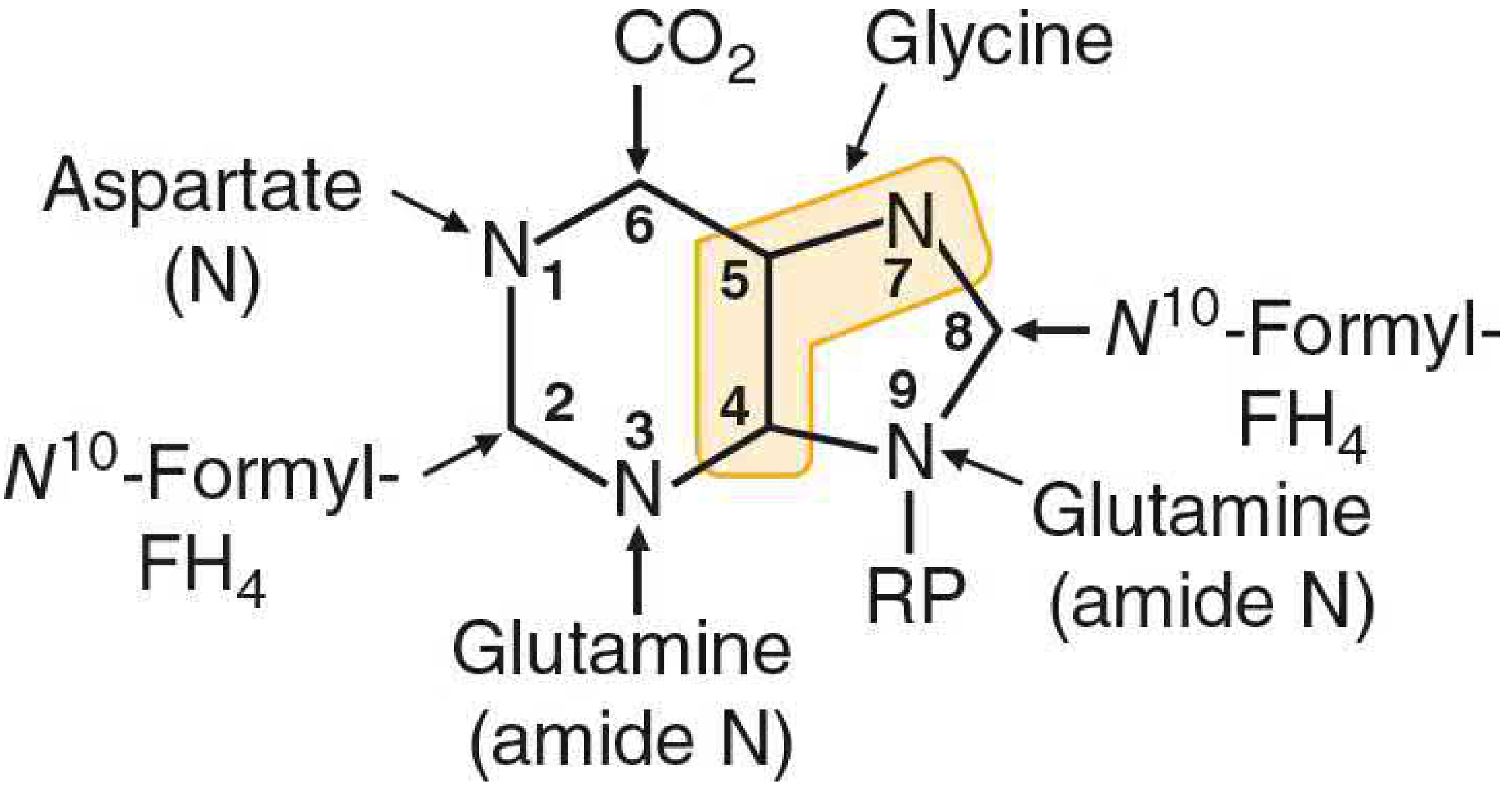
FIGURE: Origin of atoms in the purine base. Glycine (C4, C5, N7), CO2 (C6), Aspartate-N (N1), Glutamine amide-N (N3, N9), N10-formyl-FH4 (C2, C8), and RP (ribose 5'-phosphate) are the building blocks.
Key steps:
- PRPP synthetase converts ribose 5-phosphate + ATP → PRPP (rate-limiting, regulated by feedback inhibition from AMP/GMP)
- The first committed step: glutamine → PRPP → 5-phosphoribosylamine (by amidophosphoribosyltransferase)
- IMP (inosine monophosphate) is the first purine nucleotide synthesized
- IMP → AMP (via adenylosuccinate, requiring GTP as energy)
- IMP → GMP (via XMP, requiring ATP as energy)
This cross-regulation ensures balanced production of adenine and guanine nucleotides.
Regulation: Four key enzymes are allosterically regulated:
-
PRPP synthetase (inhibited by AMP, GMP, IMP)
-
Amidophosphoribosyltransferase (inhibited by AMP + GMP together)
-
IMP dehydrogenase
-
Adenylosuccinate synthetase
-
Basic Medical Biochemistry, 6e, p. 1404-1406; Harper's Illustrated Biochemistry, 32e
B. Purine Salvage Pathway
Since de novo synthesis is expensive, cells recycle free purine bases via salvage enzymes:
| Enzyme | Reaction | Clinical relevance |
|---|---|---|
| HGPRT (hypoxanthine-guanine phosphoribosyltransferase) | Hypoxanthine/guanine + PRPP → IMP/GMP | Deficiency = Lesch-Nyhan syndrome |
| APRT (adenine phosphoribosyltransferase) | Adenine + PRPP → AMP | Deficiency = 2,8-dihydroxyadenine urolithiasis |
| Adenosine kinase | Adenosine + ATP → AMP + ADP | - |
| Adenosine deaminase (ADA) | Adenosine → Inosine | Deficiency = SCID |
| Purine nucleoside phosphorylase (PNP) | Inosine/guanosine → hypoxanthine/guanine | Deficiency = T-cell immunodeficiency |
The purine nucleotide cycle in muscle converts aspartate carbons to fumarate (replenishing TCA cycle) and releases ammonia during exercise.
- Basic Medical Biochemistry, 6e, p. 1405
C. Purine Catabolism
Purine nucleosides are degraded to their free bases and then catabolized to uric acid, the end product in humans (humans lack uricase, unlike most mammals which convert uric acid to allantoin, a more water-soluble product):
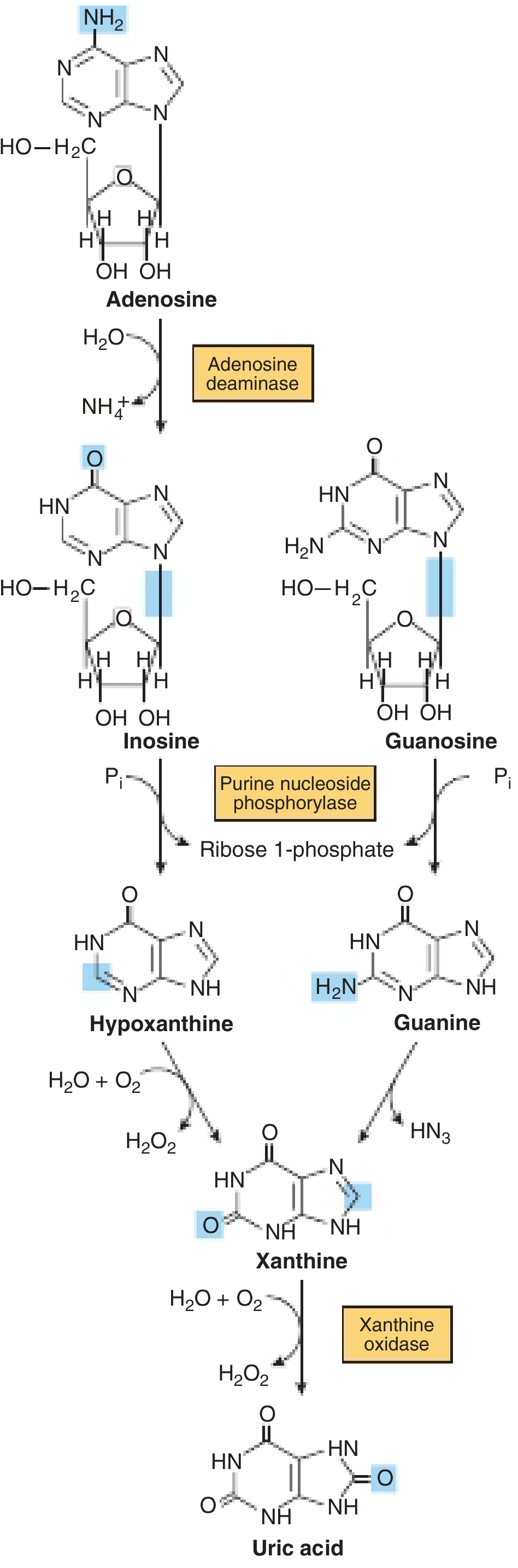
FIGURE: Formation of uric acid from purine nucleosides. Adenosine deaminase converts adenosine → inosine. Purine nucleoside phosphorylase cleaves inosine/guanosine to hypoxanthine/guanine. Xanthine oxidase converts hypoxanthine → xanthine → uric acid.
Key point: Uric acid has limited solubility. Elevated serum urate → crystal deposition in cooler peripheral tissues (e.g., first metatarsophalangeal joint) → gout.
3. Pyrimidine Metabolism
A. De Novo Synthesis
Unlike purines (assembled on ribose phosphate), pyrimidines are synthesized as the free base first, then attached to ribose phosphate.
Precursors: Aspartate + carbamoyl phosphate form all ring atoms.
Steps:
- Carbamoyl phosphate synthetase II (CPSII) - cytoplasmic enzyme using glutamine (not ammonia); forms carbamoyl phosphate. This is the first committed, rate-limiting step, regulated by UTP (inhibitor) and PRPP (activator).
- Carbamoyl phosphate + aspartate → carbamoyl aspartate → dihydroorotate → orotic acid
- Orotic acid + PRPP → OMP (orotidine monophosphate) via orotate phosphoribosyltransferase
- OMP → UMP via OMP decarboxylase (steps 3+4 are catalyzed by the bifunctional enzyme UMP synthase)
- UMP → UDP → UTP → CTP (CTP synthetase adds amino group from glutamine)
- UDP → dUDP → dUMP → dTMP (thymidylate synthase, requires N5,N10-methylene-FH4)
Key distinction from purine synthesis: Pyrimidine synthesis begins in the cytoplasm with carbamoyl phosphate made by CPSII; urea cycle uses mitochondrial carbamoyl phosphate from CPSI.
- Basic Medical Biochemistry, 6e, p. 1406; Harper's Illustrated Biochemistry, 32e
B. Deoxyribonucleotide Synthesis
Ribonucleotide reductase reduces the 2'-OH of ribonucleotide diphosphates (ADP, GDP, CDP, UDP) to deoxyribonucleotides. Two allosteric sites:
-
Activity site (ATP activates, dATP inhibits overall activity)
-
Specificity site (determines which substrate is reduced)
-
Basic Medical Biochemistry, 6e, p. 1406-1407
C. Pyrimidine Catabolism
Unlike purines, pyrimidine catabolism produces water-soluble end products: CO2, NH3, β-alanine (from uracil/cytosine), and β-aminoisobutyrate (from thymine). This means excess pyrimidine catabolism rarely causes clinical disease.
Note: Increased β-aminoisobutyrate excretion occurs in leukemia, severe radiation exposure, and in many people of East Asian ancestry (benign polymorphism).
4. Disorders of Purine Metabolism
A. Gout and Hyperuricemia
The most common purine disorder. Uric acid crystals deposit in joints and soft tissues when serum urate exceeds solubility (~6.8 mg/dL).
Causes:
- Primary (overproduction):
- PRPP synthetase overactivity (gain-of-function mutations: elevated Vmax, increased ribose 5-phosphate affinity, resistance to feedback inhibition)
- HGPRT partial deficiency (Kelley-Seegmiller syndrome)
- Secondary overproduction: Cancer, psoriasis, myeloproliferative disorders (increased cell turnover)
- Underexcretion (most common, ~90% of cases): Renal tubular handling abnormalities
- Von Gierke Disease (glucose-6-phosphatase deficiency): Enhanced ribose 5-phosphate → PRPP → purine overproduction; associated lactic acidosis raises the renal threshold for urate
Treatment:
- Acute attack: Colchicine (inhibits microtubule polymerization, blocks neutrophil migration), NSAIDs, corticosteroids
- Chronic prevention: Allopurinol (xanthine oxidase inhibitor, structural analog of hypoxanthine; reduced to alloxanthine which is a tight-binding inhibitor), febuxostat (non-purine xanthine oxidase inhibitor), uricosurics (probenecid)
B. Lesch-Nyhan Syndrome
- Defect: Complete absence of HGPRT (X-linked recessive, gene on Xq26-27)
- Mechanism: HGPRT deficiency → cannot salvage hypoxanthine/guanine → intracellular PRPP accumulates → drives de novo purine overproduction → massive hyperuricemia
- Clinical features:
- Hyperuricemia + uric acid nephrolithiasis
- Severe neurological: Self-mutilation (lip/finger biting - pathognomonic), choreoathetosis, dysarthria, hyperreflexia, hypertonia, cognitive impairment
- Behavioral: Compulsive self-injurious behavior
- Genetics: Mutations include deletions, frameshift, base substitutions, aberrant mRNA splicing
Kelley-Seegmiller syndrome = partial HGPRT deficiency: hyperuricemia + gout without the neurological features.
- Harper's, 32e, p. 355; Bradley and Daroff's Neurology, p. 1950
C. Adenosine Deaminase (ADA) Deficiency
- Defect: ADA deficiency (autosomal recessive; most common cause of autosomal recessive SCID)
- Mechanism: ADA normally converts adenosine → inosine and deoxyadenosine → deoxyinosine. Deficiency → accumulation of deoxyadenosine → accumulates as dATP in lymphocytes (lymphocytes lack efficient dephosphorylation) → dATP inhibits ribonucleotide reductase → depletes DNA precursors → lymphocyte apoptosis
- Clinical: Severe Combined Immunodeficiency (SCID) - absent T cells AND B cells; infants die from infections without treatment
- Treatment: Enzyme replacement therapy (PEG-ADA), gene therapy (historically important first successful gene therapy), bone marrow transplant
D. Purine Nucleoside Phosphorylase (PNP) Deficiency
- Defect: PNP (autosomal recessive)
- Mechanism: Accumulation of dGTP → inhibits ribonucleotide reductase → T cell depletion
- Clinical: Severe T-cell deficiency with apparently normal B-cell function; autoimmune disorders; benign and opportunistic infections
- Contrast with ADA deficiency: PNP affects mainly T cells; ADA affects both T and B cells
E. Xanthinuria (Xanthine Oxidase Deficiency)
- Defect: Xanthine oxidase (genetic or severe liver damage)
- Mechanism: Cannot convert hypoxanthine/xanthine → uric acid
- Clinical: Hypouricemia + increased urinary excretion of hypoxanthine and xanthine; severe cases → xanthine urolithiasis (xanthine stones are less soluble than uric acid)
5. Disorders of Pyrimidine Metabolism
A. Orotic Aciduria
Type I - deficiency of both UMP synthase activities (orotate phosphoribosyltransferase + OMP decarboxylase):
- Orotic acid accumulates and is excreted in urine
- Megaloblastic anemia (cannot synthesize pyrimidines → no DNA synthesis in erythroid precursors)
- Does NOT respond to folate or B12
- Treatment: Uridine supplementation (bypasses the block; UMP is used downstream)
Type II - deficiency of OMP decarboxylase alone (rarer)
Secondary orotic aciduria: Occurs in urea cycle defects (especially OTC deficiency) where excess carbamoyl phosphate overflows to pyrimidine synthesis; also occurs with allopurinol/6-azauridine therapy.
B. Dihydropyrimidine Dehydrogenase (DPD) Deficiency
- Defect: DPD (autosomal recessive)
- Pathway role: DPD catalyzes the first step of pyrimidine catabolism: uracil → dihydrouracil; thymine → dihydrothymine
- Clinical genetics: Often asymptomatic; neurological complications in severe cases (intellectual disability, seizures, abnormal MRI)
- Critical pharmacogenomic significance: 5-fluorouracil (5-FU) is a substrate for DPD. Patients with DPD deficiency (even heterozygotes) have markedly reduced 5-FU metabolism → severe, life-threatening 5-FU toxicity (mucositis, myelosuppression, neurotoxicity)
- DPD testing is now recommended before 5-FU-based chemotherapy
C. Pyrimidine-5'-Nucleotidase-1 (P5'N-1) Deficiency
-
Most frequent disorder of red cell nucleotide metabolism
-
P5'N-1 dephosphorylates pyrimidine nucleotides → nucleosides → diffuse out of RBCs
-
Deficiency → pyrimidine nucleotide accumulation → precipitation in RBCs → shortened RBC lifespan
-
Clinical: Nonspherocytic hemolytic anemia; prominent basophilic stippling (pathognomonic - the only RBC enzyme deficiency where morphology is diagnostic)
-
Inheritance: Autosomal recessive; gene NT5C3A on chromosome 7p14.3
-
Acquired form: Lead poisoning inhibits P5'N-1 → explains basophilic stippling in lead toxicity
-
Tietz Textbook of Laboratory Medicine, 7e, p. 1856
6. Summary Table: Purine & Pyrimidine Disorders
| Disorder | Defective Enzyme | Key Feature | Clinical Presentation |
|---|---|---|---|
| Gout | PRPP synthetase (overactive) or renal handling | Uric acid crystals | Painful arthritis, tophi, nephrolithiasis |
| Lesch-Nyhan | HGPRT (complete) | X-linked recessive; PRPP excess | Self-mutilation, choreoathetosis, hyperuricemia |
| Kelley-Seegmiller | HGPRT (partial) | Partial HGPRT deficiency | Gout, no neurological features |
| ADA deficiency | Adenosine deaminase | dATP accumulation | SCID (T + B cell deficiency) |
| PNP deficiency | Purine nucleoside phosphorylase | dGTP accumulation | T-cell immunodeficiency, normal B cells |
| Von Gierke | Glucose-6-phosphatase | Excess PRPP | Secondary gout + hypoglycemia, lactic acidosis |
| Xanthinuria | Xanthine oxidase | Cannot make uric acid | Hypouricemia, xanthine stones |
| Orotic aciduria I | UMP synthase | Orotic acid excess | Megaloblastic anemia; responds to uridine |
| DPD deficiency | Dihydropyrimidine dehydrogenase | 5-FU toxicity | Neurological; 5-FU pharmacotoxicity |
| P5'N-1 deficiency | Pyrimidine-5'-nucleotidase-1 | Pyrimidine accumulation in RBCs | Hemolytic anemia + basophilic stippling |
7. Pharmacological Targets in This Pathway
| Drug | Target | Use |
|---|---|---|
| Allopurinol | Xanthine oxidase (competitive/irreversible via alloxanthine) | Gout, tumor lysis syndrome |
| Febuxostat | Xanthine oxidase (non-purine inhibitor) | Gout (fewer drug interactions) |
| Methotrexate | Dihydrofolate reductase | Blocks N10-formyl-FH4 → blocks both purine and thymidylate synthesis |
| 5-Fluorouracil | Thymidylate synthase (via FdUMP) | Cancer; requires DPD for catabolism |
| Hydroxyurea | Ribonucleotide reductase | Cancer, sickle cell disease |
| 6-Mercaptopurine / 6-TG | Multiple purine synthesis steps (via HGPRT conversion to nucleotides) | Leukemia |
| Probenecid | Renal urate transporter (URAT1) | Uricosuric in gout |
Sources:
- Basic Medical Biochemistry, 6e, Chapter 39 (pp. 1404-1415) - most comprehensive pathway coverage
- Harper's Illustrated Biochemistry, 32e, Chapter 33 (pp. 354-358) - disorders table and catabolism
- Bradley and Daroff's Neurology in Clinical Practice, Chapter 91 (p. 1950) - neurological manifestations
- Tietz Textbook of Laboratory Medicine, 7e, Chapter on RBC nucleotide metabolism (pp. 1856-1860) - P5'N-1 deficiency
Disorders of purine metabolism
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
I now have comprehensive, multi-textbook content with rich images. Let me compose the full answer.
Disorders of Purine Metabolism
Purine metabolism disorders arise from disruption of de novo synthesis, salvage pathways, or catabolic pathways. Because uric acid - the end product of human purine catabolism - has poor water solubility, most purine disorders manifest as hyperuricemia, crystal deposition, or immunodeficiency (when salvage enzymes in lymphocytes are impaired).
Quick Reference: Purine Catabolism Pathway
Purine nucleoside catabolism. Adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) are critical enzymes - their deficiency causes immunodeficiency. Xanthine oxidase converts hypoxanthine/xanthine → uric acid; its inhibition is the target of allopurinol.
Purine Salvage Pathway - The Site of Most Disorders
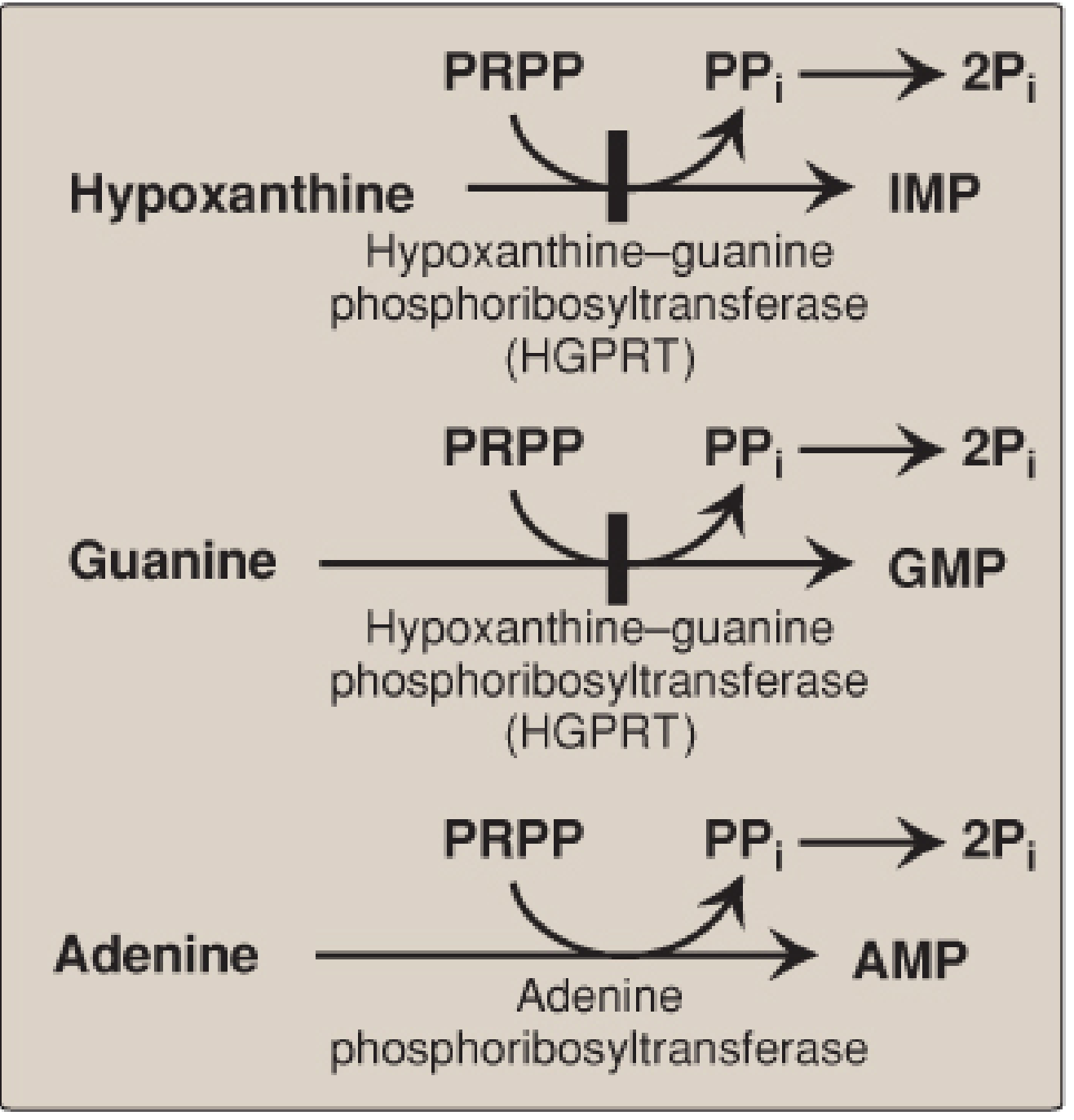
Purine salvage enzymes. HGPRT salvages hypoxanthine and guanine. APRT salvages adenine. Deficiency of HGPRT causes Lesch-Nyhan syndrome. Note the block symbol at HGPRT.
1. Gout and Hyperuricemia
Pathophysiology
Normal serum urate solubility limit is ~6.8 mg/dL. When exceeded, monosodium urate (MSU) crystals precipitate preferentially in cooler peripheral tissues (first metatarsophalangeal joint = "podagra"). Only ~10% of individuals with hyperuricemia actually develop gout.
Causes of hyperuricemia:
| Category | Mechanism | Examples |
|---|---|---|
| Overproduction | Increased purine synthesis/catabolism | PRPP synthetase overactivity, HGPRT deficiency, myeloproliferative disorders, tumor lysis |
| Underexcretion (~90% of cases) | Impaired renal urate handling | Idiopathic; thiazide diuretics; lead nephropathy; cyclosporine |
| Combined | Both | Von Gierke disease, Lesch-Nyhan syndrome |
Specific enzyme defects causing overproduction:
- PRPP synthetase gain-of-function mutations: Elevated Vmax, increased affinity for ribose 5-phosphate, or resistance to feedback inhibition by AMP/GMP - all drive excess PRPP → excess purine synthesis → excess uric acid
- HGPRT partial deficiency (Kelley-Seegmiller syndrome): Cannot salvage hypoxanthine/guanine → PRPP accumulates → de novo purine overproduction
Inflammatory Mechanism (Gout Attack)
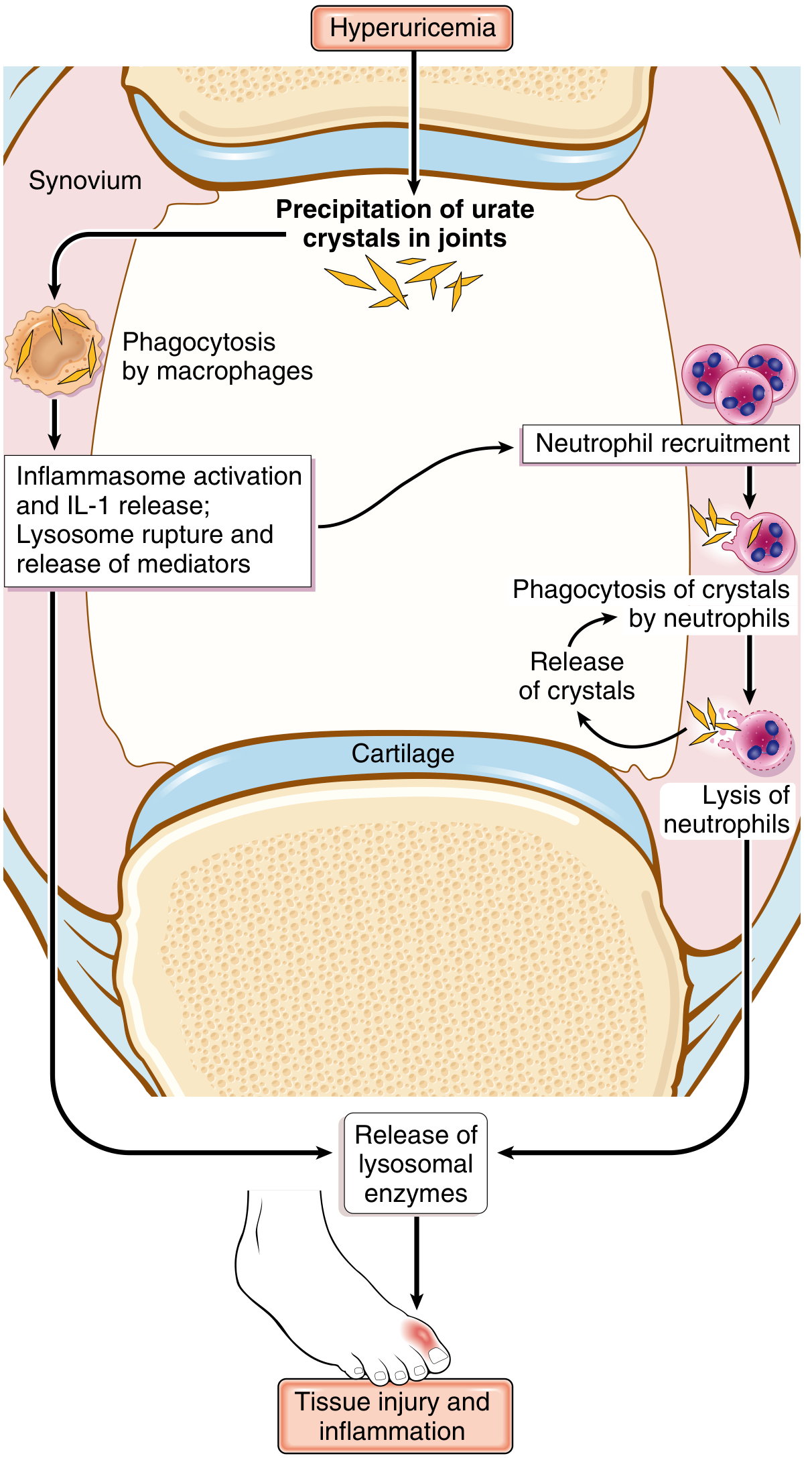
Key steps: MSU crystals → NLRP3 inflammasome activation in macrophages → IL-1β release → massive neutrophil influx → neutrophils phagocytose crystals → lysosomal rupture → proteolytic enzyme release → tissue injury.
Morphology
- Acute arthritis: Dense neutrophilic infiltrates; needle-shaped urate crystals in synovium and neutrophil cytoplasm
- Polarized light microscopy: Crystals show negative birefringence (yellow when parallel, blue when perpendicular to polarizer axis)
- Tophi: Chalky deposits of urate crystals surrounded by reactive fibroblasts, mononuclear cells, and giant cells; erode cartilage and bone
- Gouty nephropathy: Urate crystal deposition in renal interstitium; can cause chronic kidney disease; acute uric acid nephropathy causes AKI (especially in tumor lysis syndrome)
Treatment
| Drug | Mechanism | Use |
|---|---|---|
| Colchicine | Inhibits microtubule polymerization; blocks neutrophil migration and inflammasome activation | Acute attack + prophylaxis |
| NSAIDs (indomethacin) | COX inhibition | Acute attack |
| Corticosteroids | Broad anti-inflammatory | Acute attack (when NSAIDs/colchicine contraindicated) |
| Allopurinol | Xanthine oxidase inhibitor (structural analogue of hypoxanthine; oxidized to alloxanthine, which tightly inhibits the enzyme) | Chronic urate lowering |
| Febuxostat | Non-purine xanthine oxidase inhibitor | Chronic urate lowering; fewer drug interactions than allopurinol |
| Probenecid | Blocks URAT1 (renal urate reabsorption transporter) | Uricosuric; only when underexcretor |
| Rasburicase | Recombinant uricase; converts uric acid → allantoin (water-soluble) | Tumor lysis syndrome prophylaxis/treatment |
| Pegloticase | PEGylated uricase | Refractory chronic gout |
- Robbins Pathologic Basis of Disease, pp. 1112-1114; Harper's Illustrated Biochemistry, 32e, p. 354
2. Lesch-Nyhan Syndrome
The prototypical purine salvage disorder.
| Feature | Detail |
|---|---|
| Gene | HPRT1 (Xq26-27) |
| Inheritance | X-linked recessive (affects males; females are carriers) |
| Enzyme | Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) - complete deficiency |
Biochemical Mechanism
HGPRT salvages hypoxanthine → IMP and guanine → GMP. With complete deficiency:
- Hypoxanthine and guanine cannot be salvaged → both are catabolized to uric acid (massive overproduction)
- Intracellular PRPP accumulates (no longer consumed by salvage) → drives de novo purine overproduction further
- IMP and GMP are depleted → loss of feedback inhibition of amidophosphoribosyltransferase (GPAT) → de novo synthesis runs unchecked
Mutations include deletions, frameshift mutations, base substitutions, and aberrant mRNA splicing.
Clinical Features
"The 3 H's":
- Hyperuricemia - uric acid nephrolithiasis, gouty arthritis, tophi (can appear in first weeks of life as orange "uric acid sand" in diapers)
- Hypotonia progressing to hypertonia, choreoathetosis, spasticity, dysarthria, hyperreflexia
- Harm (self-mutilation) - pathognomonic compulsive self-injurious behavior: biting of lips, fingers, and buccal mucosa; paradoxically patients are distressed by their own behavior
Other features: cognitive impairment, megaloblastic anemia (from purine depletion in rapidly dividing cells).
Kelley-Seegmiller syndrome = partial HGPRT deficiency: severe gout ± mild neurological features, but no self-mutilation. As functional enzyme increases, severity decreases.
Treatment
- Allopurinol controls hyperuricemia and prevents nephropathy/gout
- No treatment reverses the neurological features - the most disabling aspect
- Physical restraints are often used to prevent self-injury (paradoxically, many patients request them)
- Symptomatic: baclofen, benzodiazepines for spasticity/dystonia
- Harper's 32e, p. 355-356; Lippincott Biochemistry 8e, pp. 834-835; Tietz Laboratory Medicine, 7e
3. Adenosine Deaminase (ADA) Deficiency → SCID
Biochemistry
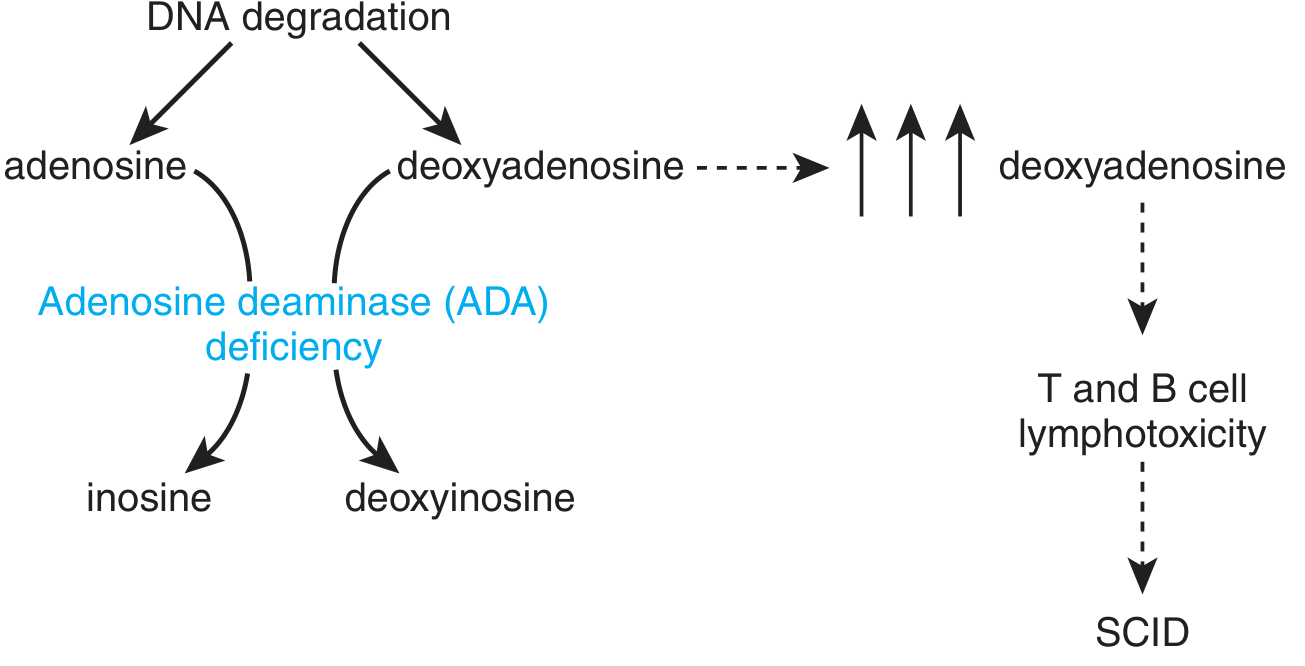
ADA deficiency: deoxyadenosine accumulates and is phosphorylated to dATP in lymphocytes. dATP inhibits ribonucleotide reductase, blocking DNA synthesis and triggering apoptosis of lymphocyte progenitors.
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive |
| Frequency | ~15-20% of all SCID cases; most common cause of autosomal recessive SCID |
| Enzyme | Adenosine deaminase: adenosine → inosine; deoxyadenosine → deoxyinosine |
Mechanism
- ADA deficiency → deoxyadenosine accumulates
- Lymphocytes (especially T cells) lack efficient dephosphorylation enzymes → deoxyadenosine is phosphorylated to dATP
- dATP directly inhibits ribonucleotide reductase → DNA precursor depletion → lymphocyte apoptosis
- Both T cells and B cells are decimated (though T cells are more severely affected)
- Extra-lymphoid effects: Bone dysplasia with abnormal costochondral junctions and metaphyses (50% of cases); neurologic defects; because ADA is a ubiquitous enzyme
Clinical Features
- Severe Combined Immunodeficiency (SCID): Absent T cells, B cells, and NK cells
- Onset in early infancy with recurrent, life-threatening infections (bacterial, viral, fungal, opportunistic)
- Failure to thrive
- Thymus is small and devoid of lymphocytes (vs. X-linked SCID where thymic lobules retain epithelial structure)
- Without treatment, fatal within the first 1-2 years of life
Treatment
- Hematopoietic stem cell transplantation (HSCT) - curative standard of care
- PEG-ADA (pegylated bovine ADA): Enzyme replacement; modified ADA stays in extracellular fluid where it degrades toxic purines before they enter lymphocytes. PEGylation extends half-life (3-6 days) and reduces immunogenicity. Restores immunoprotection but does not fully correct T lymphopenia.
- Gene therapy: ADA deficiency was one of the first diseases treated with gene therapy (1990). Current trials use safer lentiviral vectors with excellent outcomes. ADA-SCID gene therapy is now approved (Strimvelis in Europe).
- Robbins, p. 232; Harrison's 22e, p. 2842; Thompson & Thompson Genetics, p. 319-320
4. Purine Nucleoside Phosphorylase (PNP) Deficiency
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive |
| Enzyme | Purine nucleoside phosphorylase: inosine/guanosine → hypoxanthine/guanine + ribose 1-phosphate |
| Frequency | Very rare; <100 cases reported worldwide |
Mechanism
- PNP deficiency → accumulation of inosine, guanosine, deoxyinosine, and especially deoxyguanosine
- dGTP accumulates specifically in T cells (T cells lack efficient dephosphorylation)
- dGTP inhibits ribonucleotide reductase → T cell apoptosis
- B cells have efficient pathways to dephosphorylate dGTP → B cells are relatively spared
Clinical Features
- Selective profound T-cell deficiency with apparently normal B-cell numbers and function
- Recurrent infections, particularly viral and opportunistic
- Autoimmune manifestations (due to abnormal immune regulation without adequate T-cell suppression)
- Severe neurologic impairments (intellectual disability, spastic diplegia, cerebellar ataxia) - important distinguishing feature from ADA deficiency
- Hypouricemia (cannot produce uric acid from inosine/guanosine)
Contrast with ADA deficiency:
| Feature | ADA Deficiency | PNP Deficiency |
|---|---|---|
| Cells affected | T + B + NK | Mainly T cells |
| Toxic metabolite | dATP | dGTP |
| Uric acid | Normal or elevated | Low (hypouricemia) |
| Neurologic features | Moderate | Prominent |
| Autoimmune features | Less common | Common |
- Harper's 32e, p. 357; Harrison's 22e, p. 2842; Bradley & Daroff Neurology
5. Von Gierke Disease (Glycogen Storage Disease Type 1a) - Secondary Hyperuricemia
| Feature | Detail |
|---|---|
| Primary defect | Glucose-6-phosphatase deficiency |
| Mechanism of hyperuricemia | Enhanced glycolysis with pentose phosphate pathway shunting → excess ribose 5-phosphate → excess PRPP → overproduction of purines |
| Compounding factor | Associated lactic acidosis competes with uric acid for renal tubular secretion → elevates renal threshold for urate → further raises serum uric acid |
| Clinical | Severe hypoglycemia, hepatomegaly, growth retardation + secondary gout |
- Harper's 32e, p. 356
6. Xanthinuria (Xanthine Oxidase Deficiency)
| Feature | Detail |
|---|---|
| Defect | Xanthine oxidase (genetic or acquired from severe liver disease) |
| Consequence | Cannot convert hypoxanthine/xanthine → uric acid |
| Lab findings | Hypouricemia; elevated urinary xanthine and hypoxanthine |
| Clinical | Often asymptomatic; severe cases → xanthine urolithiasis (xanthine stones; less soluble than uric acid at physiologic urinary pH) |
| Molybdenum cofactor deficiency | Combined deficiency of xanthine oxidase + sulfite oxidase (both require Mo cofactor); causes severe neurological disease in neonates |
7. Adenine Phosphoribosyltransferase (APRT) Deficiency
| Feature | Detail |
|---|---|
| Defect | APRT (salvages adenine → AMP) |
| Inheritance | Autosomal recessive |
| Consequence | Adenine cannot be salvaged → adenine is oxidized by xanthine oxidase to 2,8-dihydroxyadenine (2,8-DHA) |
| Clinical | 2,8-DHA urolithiasis (radiolucent stones, often misidentified as uric acid stones); can cause acute and chronic kidney disease |
| Uric acid levels | Normal (uric acid pathway unaffected) |
| Diagnosis | Stone analysis; urine microscopy shows distinctive brown crystals; genetic testing |
| Treatment | Allopurinol (inhibits xanthine oxidase, blocking 2,8-DHA production) + low-purine diet |
- Campbell-Walsh Urology
8. PRPP Synthetase Superactivity
| Feature | Detail |
|---|---|
| Defect | Gain-of-function mutations in PRPP synthetase |
| Inheritance | X-linked |
| Mechanism | Elevated Vmax; increased affinity for ribose 5-phosphate; resistance to feedback inhibition by AMP/GMP → excess PRPP → overproduction of purines |
| Clinical | Early-onset severe gout, nephrolithiasis; some variants associated with sensorineural deafness and neurodevelopmental features |
9. Comprehensive Summary Table
| Disorder | Defective Enzyme | Pathway | Inheritance | Key Biochemical Finding | Cardinal Clinical Feature |
|---|---|---|---|---|---|
| Gout (primary) | PRPP synthetase (gain-of-function) / renal handling | Synthesis / excretion | X-linked / polygenic | Hyperuricemia | Painful arthritis, tophi, nephropathy |
| Lesch-Nyhan syndrome | HGPRT (complete) | Salvage | X-linked recessive | ↑↑ Uric acid, ↑ PRPP | Self-mutilation, choreoathetosis, hyperuricemia |
| Kelley-Seegmiller | HGPRT (partial) | Salvage | X-linked recessive | ↑ Uric acid | Gout only; no neurological features |
| ADA deficiency | Adenosine deaminase | Catabolism | Autosomal recessive | ↑ dATP in lymphocytes | SCID (T + B + NK cell deficiency) |
| PNP deficiency | Purine nucleoside phosphorylase | Catabolism | Autosomal recessive | ↑ dGTP; hypouricemia | T-cell SCID + neurological impairment |
| Von Gierke disease | Glucose-6-phosphatase | Secondary (glycogen storage) | Autosomal recessive | ↑ PRPP, lactic acidosis | Hypoglycemia + secondary gout |
| Xanthinuria | Xanthine oxidase | Catabolism | Autosomal recessive | Hypouricemia, ↑ urinary xanthine | Xanthine urolithiasis |
| APRT deficiency | APRT | Salvage | Autosomal recessive | ↑ 2,8-DHA in urine | 2,8-DHA kidney stones; normal uric acid |
| PRPP synthetase superactivity | PRPP synthetase | Synthesis | X-linked | ↑↑ PRPP, ↑ uric acid | Early severe gout ± deafness |
10. Key Pharmacological Targets in Purine Disorders
| Drug | Target | Clinical Use |
|---|---|---|
| Allopurinol | Xanthine oxidase (inhibited by alloxanthine) | Gout, Lesch-Nyhan, APRT deficiency, tumor lysis prophylaxis |
| Febuxostat | Xanthine oxidase (non-purine inhibitor) | Gout (fewer drug interactions) |
| Rasburicase / Pegloticase | Uricase (converts uric acid → allantoin) | Tumor lysis / refractory gout |
| Colchicine | Microtubule polymerization; NLRP3 inflammasome | Acute gout; prophylaxis |
| PEG-ADA | Exogenous ADA enzyme replacement | ADA-SCID |
| 6-Mercaptopurine / Azathioprine | Converted by HGPRT to cytotoxic nucleotides | Leukemia; immunosuppression (note: requires HGPRT) |
| Methotrexate | Dihydrofolate reductase | Blocks N10-formyl-FH4 → impairs de novo purine synthesis |
Sources:
- Harper's Illustrated Biochemistry, 32e, Ch. 33, pp. 354-357 - disorders table, Lesch-Nyhan, Von Gierke, ADA/PNP
- Lippincott Illustrated Reviews: Biochemistry, 8e, Ch. 22, pp. 834-835 - Lesch-Nyhan mechanism and salvage pathway diagram
- Robbins & Cotran Pathologic Basis of Disease, Ch. 6 and Ch. 26, pp. 232, 1112-1114 - ADA/SCID pathology; gout morphology and inflammatory mechanism
- Harrison's Principles of Internal Medicine, 22e, p. 2842 - ADA/PNP deficiency in SCID classification
- Thompson & Thompson Genetics and Genomics in Medicine, 9e, pp. 319-320 - PEG-ADA and gene therapy for ADA-SCID
- Basic Medical Biochemistry, 6e, Ch. 39, pp. 1404-1415 - comprehensive pathway overview
This is a shared conversation. Sign in to Orris to start your own chat.