Explain the concept of cor pulmonale in COPD
Reading File
Finding Sources
Finding Sources
Searching PubMed
"cor pulmonale" AND "COPD"
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I have comprehensive information from multiple authoritative textbooks along with excellent diagrams. I can now compile a thorough answer.
Cor Pulmonale in COPD
Cor pulmonale is defined as right ventricular (RV) hypertrophy and dilation - frequently accompanied by right-sided heart failure - caused by pulmonary hypertension (PH) attributable to primary disorders of the lung parenchyma or pulmonary vasculature. The term was coined by Paul Dudley White nearly a century ago. Critically, RV changes secondary to left heart disease or congenital heart disease are excluded by definition.
In COPD, cor pulmonale is a late and serious complication representing the cardiopulmonary endpoint of progressive lung destruction.
Pathogenesis Diagram
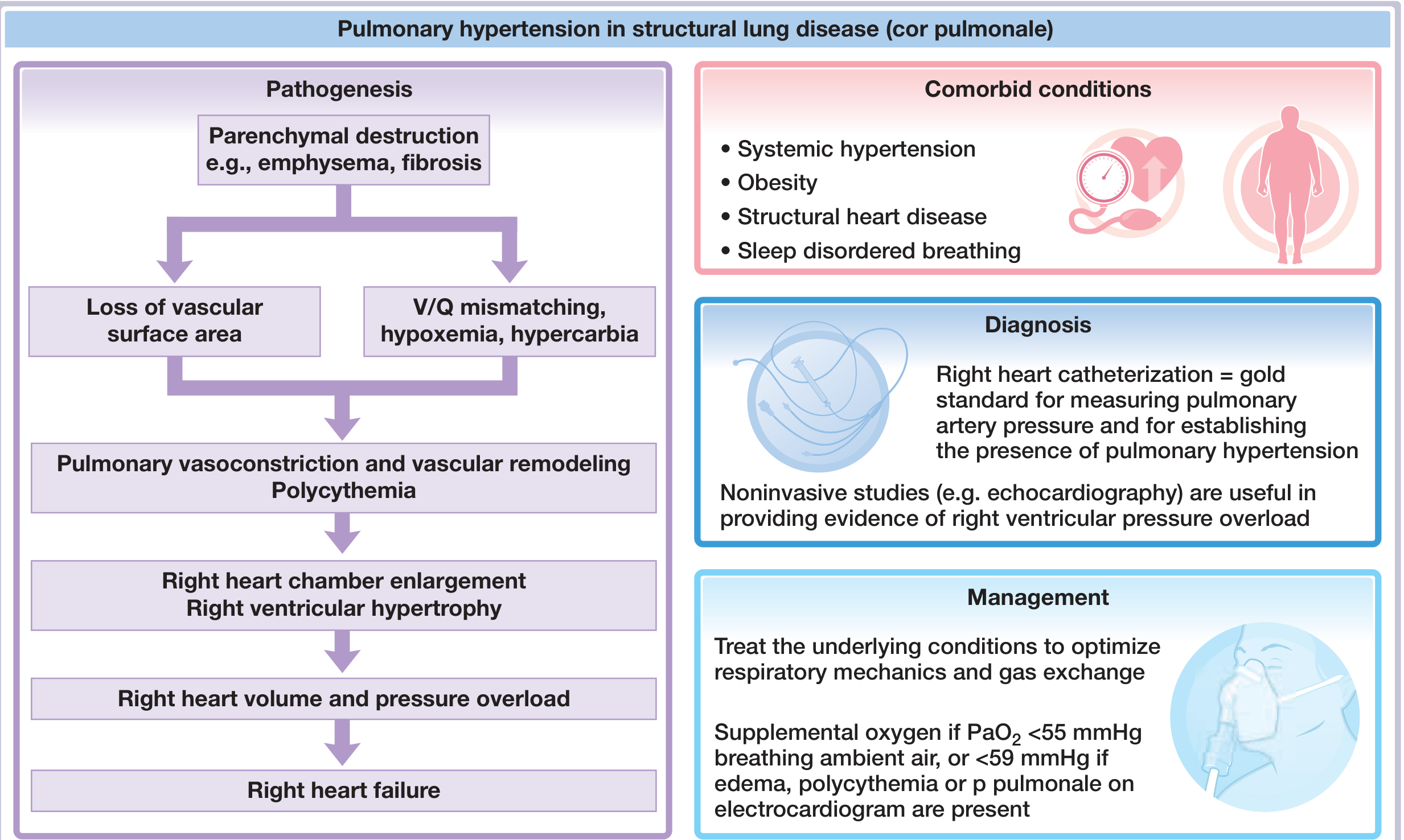
Central illustration from Fuster & Hurst's The Heart, 15th Edition - showing the full pathogenic cascade of cor pulmonale in structural lung disease.
Pathophysiology: The Cascade from COPD to Cor Pulmonale
There are several mechanisms at work, and they reinforce each other:
1. Hypoxic Pulmonary Vasoconstriction (HPV)
The dominant driver. As COPD progresses, ventilation-perfusion (V/Q) mismatch causes progressive alveolar hypoxia. The pulmonary vasculature responds to low PO2 by vasoconstriction - a reflex that is adaptive in acute settings (diverting blood from poorly ventilated alveoli) but becomes chronically damaging when hypoxia is sustained. Hypercarbia also acts independently as a potent vasoconstrictor.
2. Loss of Vascular Surface Area
Emphysematous destruction of alveolar walls obliterates the alveolar capillary bed. Normally the pulmonary circulation can compensate for loss of up to ~50% of its vascular surface area before resting pressures rise; once this reserve is exceeded, pulmonary artery pressure rises irreversibly.
3. Vascular Remodeling
Chronic hypoxia and shear stress trigger structural changes in pulmonary vessel walls: smooth muscle hypertrophy, intimal fibrosis, and endothelial dysfunction (reduced nitric oxide production, increased endothelin-1). This converts functional (reversible) vasoconstriction into fixed (irreversible) obstruction.
4. Polycythemia and Increased Blood Viscosity
Hypoxia-driven erythropoietin release causes secondary polycythemia, raising blood viscosity and further increasing pulmonary vascular resistance (PVR). This compounds the pressure burden on the RV.
5. Hyperinflation
Lung hyperinflation in COPD compresses alveolar capillaries mechanically, adding a further anatomical component to elevated PVR.
Right Ventricular Response
The RV is the downstream casualty of elevated PVR. Its response follows a sequence:
| Stage | RV Change | Hemodynamics |
|---|---|---|
| Early | RV hypertrophy (pressure overload adaptation) | Elevated PA pressure, preserved cardiac output |
| Intermediate | RV dilation (wall stress exceeds adaptive capacity) | Further PA pressure rise, normal CO at rest |
| Late | RV failure | Reduced cardiac output, elevated CVP, peripheral edema |
Only late in the course of chronic cor pulmonale does overt right heart failure with decreased cardiac output ensue. The RV is exquisitely sensitive to increases in afterload - unlike the LV, it has thin walls and limited ability to sustain high pressures chronically.
Morphologically in chronic cor pulmonale, RV wall thickness may approach or even exceed that of the LV. The RV free wall and trabeculae are prominently hypertrophied, and the RV chamber dilates as failure sets in. The shape and volume of the LV may also be distorted secondarily by the enlarged RV pushing against the interventricular septum (interventricular dependence).
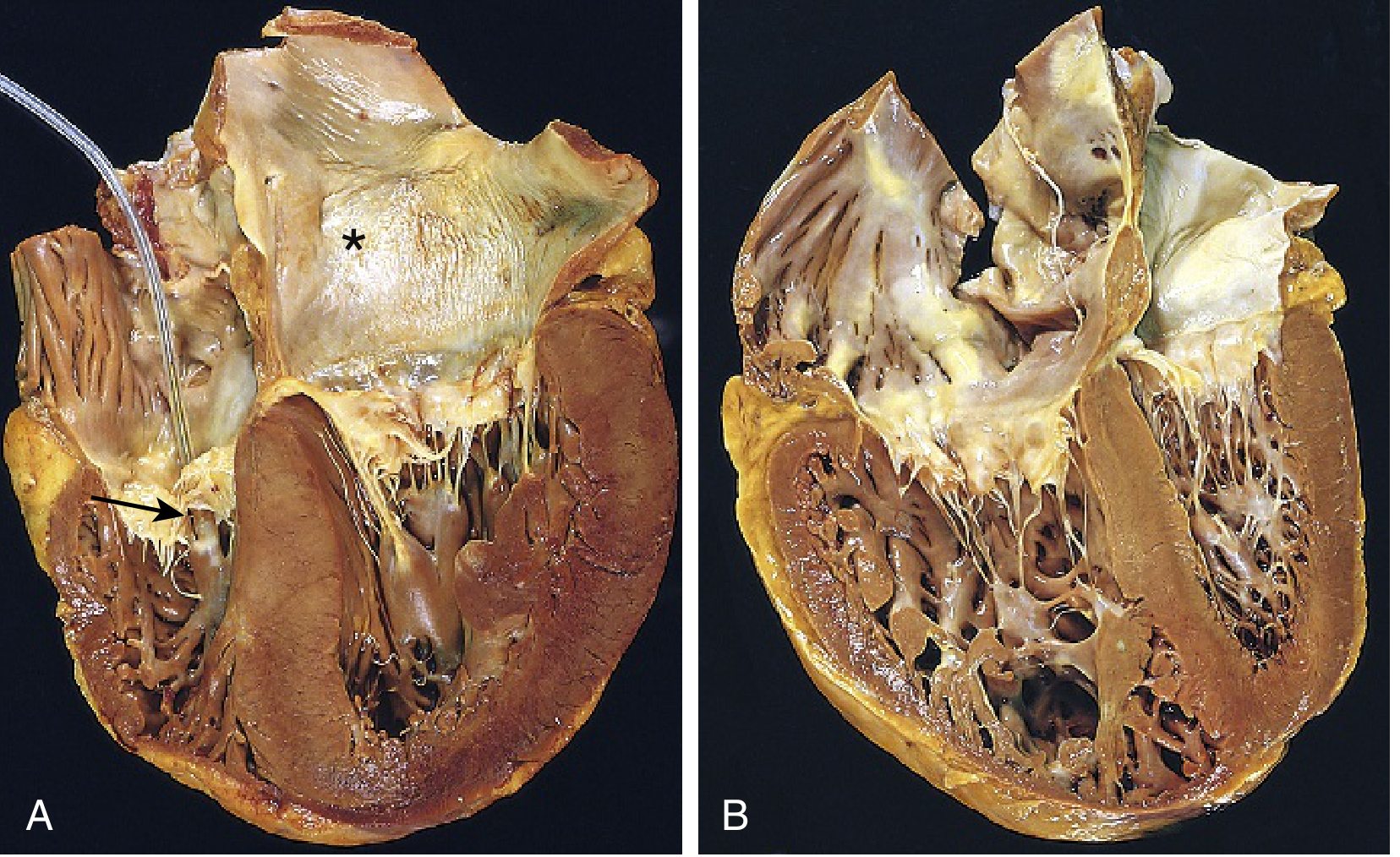
Gross pathology from Robbins & Kumar Basic Pathology. Panel B shows chronic cor pulmonale: the right ventricle is markedly dilated and hypertrophied. Note how the enlarged RV distorts the left ventricular geometry.
Clinical Manifestations
Symptoms of cor pulmonale overlap heavily with advanced COPD and are often masked by the underlying lung disease.
Symptoms:
- Exertional dyspnea (dominant, often pre-existing from COPD)
- Fatigue and reduced exercise tolerance
- Exertional syncope or near-syncope (severe PH, reduced cardiac output)
- Right upper quadrant discomfort (hepatic congestion)
Physical signs:
- Raised JVP with prominent a- or v-waves
- Right ventricular heave (parasternal lift) - difficult to detect behind a hyperinflated chest
- Loud P2 (pulmonary component of S2)
- Tricuspid regurgitation murmur (pansystolic at left sternal border, increases with inspiration)
- Peripheral pitting edema - bilateral ankle/leg edema
- Hepatomegaly and ascites in advanced right heart failure
- Central cyanosis from hypoxemia
Note: Physical findings of venous engorgement and RV hypertrophy/dilation are late signs. Peripheral edema is poorly correlated with resting right atrial pressure and may reflect RAAS activation rather than true right heart failure.
Edema in Cor Pulmonale
The mechanism of edema formation in cor pulmonale is multifactorial and not simply explained by elevated right atrial pressure alone. Hypoxemia and hypercarbia stimulate:
- Activation of the renin-angiotensin-aldosterone system (RAAS) - sodium and water retention
- Increased sympathetic tone - renal vasoconstriction reducing GFR
- Secondary hyperaldosteronism independent of RV filling pressures
This explains why peripheral edema can develop even when RV pressures are only modestly elevated, and why it may persist despite modest improvements in hemodynamics.
Investigations
ECG findings (signs of right heart strain/hypertrophy):
- P pulmonale (peaked P waves >2.5 mm in II, III, aVF - right atrial enlargement)
- Right axis deviation
- Right bundle branch block (complete or incomplete)
- R/S ratio >1 in V1 (RV hypertrophy)
- S1Q3T3 pattern (if also pulmonary embolism)
- Deep S waves in V5-V6
Echocardiography:
- RV dilation and hypertrophy
- Flattening or paradoxical motion of the interventricular septum (D-shaped LV in short axis)
- Tricuspid regurgitation - allows estimation of PA systolic pressure (PASP) by Doppler
- Note: Doppler estimates of PASP correlate poorly with invasive catheterization measurements in COPD patients (due to hyperinflation affecting acoustic windows and TR jet quality)
Chest X-ray:
- Cardiomegaly with prominent right heart border
- Enlarged main pulmonary artery and hilar vessels ("pruning" of peripheral vessels)
- Features of underlying COPD: hyperinflation, flattened diaphragms
Right Heart Catheterization (gold standard):
- Direct measurement of PA pressure (mean PAP >25 mmHg confirms PH)
- PCWP normal (<15 mmHg) - distinguishing from left heart disease
- Elevated PVR
- Reduced cardiac output in advanced disease
CT/MRI:
- CT: PA diameter >29 mm suggests PH; also shows emphysema extent
- Cardiac MRI: most accurate for RV volumes and function
Prognosis
Cor pulmonale carries a poor prognosis in COPD. Once pulmonary artery pressure exceeds 25 mmHg, average 5-year survival is reduced by approximately 50%. The onset of frank right heart failure with peripheral edema marks a critical inflection point in disease trajectory.
Management
The principles of management follow directly from the pathophysiology:
1. Treat the Underlying COPD (Primary Goal)
Optimizing respiratory mechanics and gas exchange reduces the hypoxic drive to pulmonary vasoconstriction:
- Bronchodilators (LABAs, LAMAs) - reduce air trapping and hyperinflation
- Inhaled corticosteroids - reduce exacerbations
- Smoking cessation - most important intervention at any stage
- Pulmonary rehabilitation
2. Long-Term Oxygen Therapy (LTOT) - Cornerstone
Supplemental oxygen is indicated when:
- PaO2 <55 mmHg (breathing ambient air)
- PaO2 <59 mmHg if edema, polycythemia, or P pulmonale on ECG are present
LTOT (>15 hours/day) reduces hypoxic vasoconstriction, stabilizes or mildly reduces PAP, and has been shown to improve survival in hypoxemic COPD patients. It is the only therapy proven to alter the natural history.
3. Diuretics
Used for symptomatic management of edema and volume overload (right heart failure). Spironolactone addresses the aldosterone-driven component specifically. Aggressive diuresis must be avoided as it can reduce RV preload excessively.
4. Pulmonary Vasodilators
The role of pulmonary vasodilators (calcium channel blockers, PDE-5 inhibitors, ERA, prostanoids) is not established in Group 3 PH (due to lung disease). They can worsen ventilation-perfusion mismatch by dilating vessels in poorly ventilated lung regions, worsening hypoxemia. Calcium channel blockers are generally avoided. Vasodilator therapy is reserved for selected patients with disproportionate PH (mean PAP >35 mmHg out of proportion to lung disease severity), ideally managed at specialist centers.
5. Treat Comorbidities
- Sleep-disordered breathing (OSA overlap syndrome) - CPAP/NIV
- Polycythemia - phlebotomy if hematocrit >55% causing symptoms; LTOT to address root cause
- Arrhythmias (especially supraventricular tachycardias from right atrial enlargement)
6. Advanced Therapies
- Lung transplantation - definitive treatment for end-stage COPD-related cor pulmonale
- Lung volume reduction surgery (LVRS) - selected patients with upper lobe predominant emphysema
Summary
| Aspect | Key Point |
|---|---|
| Definition | RV hypertrophy/dilation from PH due to lung disease (not left heart disease) |
| Primary driver in COPD | Chronic alveolar hypoxia → HPV → vascular remodeling |
| Additional drivers | Loss of capillary bed, polycythemia, hyperinflation, endothelial dysfunction |
| Cardinal sign | Mean PAP >25 mmHg by right heart catheterization |
| RV response | Hypertrophy → dilation → failure (sequential) |
| ECG hallmark | P pulmonale, RVH, right axis deviation |
| Best treatment | Long-term oxygen therapy + treat underlying COPD |
| Prognosis | Poor; 5-year survival reduced ~50% once PAP >25 mmHg |
Sources: Fuster & Hurst's The Heart, 15th Edition (Ch. 59); Fishman's Pulmonary Diseases and Disorders; Robbins & Kumar Basic Pathology, 10th Edition; Swanson's Family Medicine Review.
Why the pulmonary artery pressure rises in this ??
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, well-sourced answer with diagrams. Let me compile it.
Why Does Pulmonary Artery Pressure Rise in COPD?
The fundamental equation to keep in mind is:
Mean PA Pressure = Pulmonary Vascular Resistance (PVR) × Cardiac Output + Left Atrial Pressure
Since left atrial pressure is normal in cor pulmonale (no left heart disease), PA pressure rises when PVR increases and/or cardiac output rises. In COPD, multiple mechanisms drive PVR upward simultaneously and progressively.
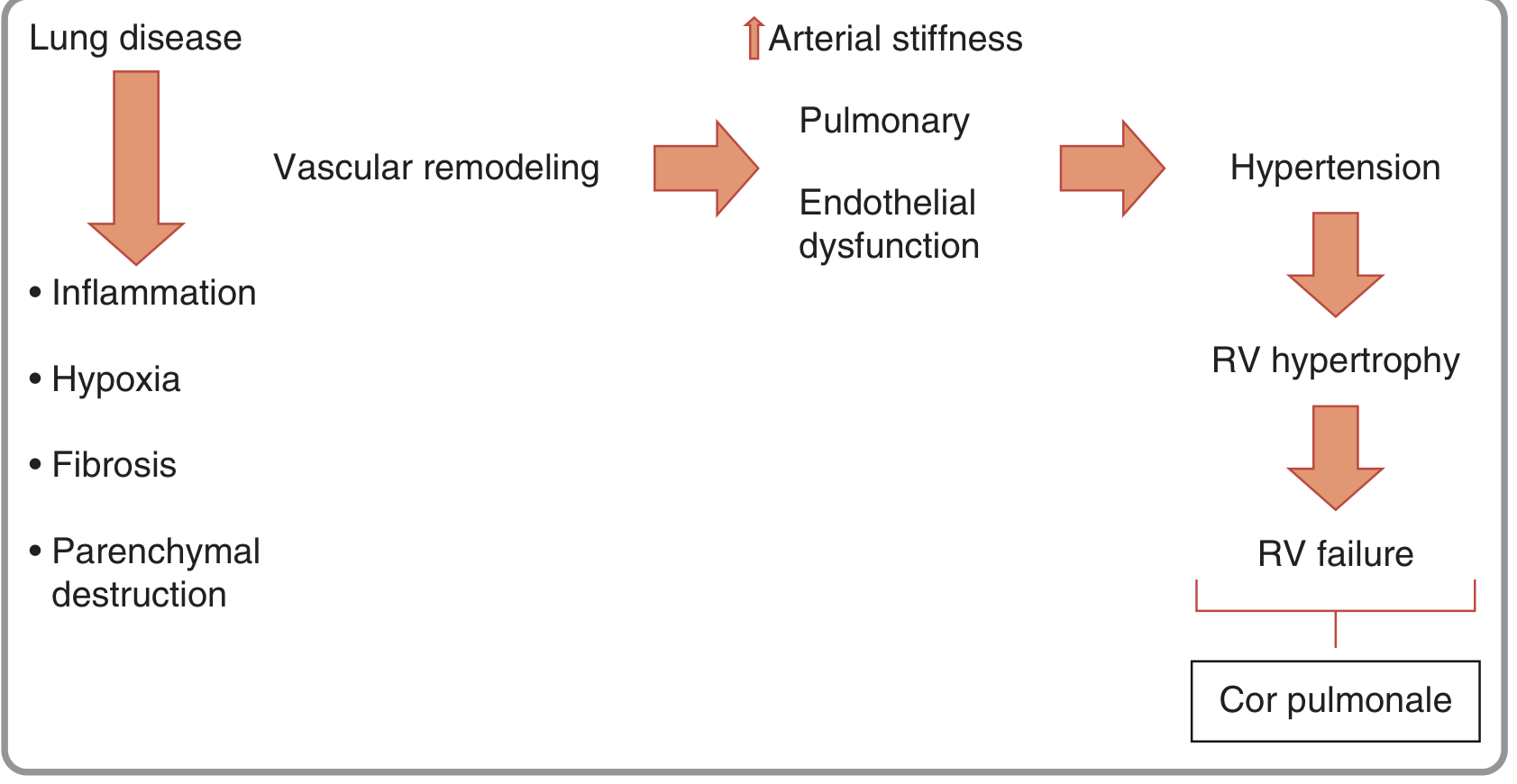
Figure from Fuster & Hurst's The Heart, 15th Edition - showing how lung disease leads to cor pulmonale through vascular remodeling and endothelial dysfunction.
Mechanism 1: Hypoxic Pulmonary Vasoconstriction (HPV) - The Dominant Driver
This is by far the most important mechanism.
The physiology
In a normal lung, hypoxic vasoconstriction is actually adaptive and local - when one region of lung is poorly ventilated (low alveolar PO2), the nearby arterioles constrict to redirect blood flow toward better-ventilated alveoli, preserving V/Q matching.
The trigger threshold: when alveolar PO2 (PAO2) falls below ~70 mmHg, pulmonary arteriolar smooth muscle senses the low O2 and constricts.
The cellular mechanism (HPV)
- Low PO2 inhibits voltage-gated K⁺ channels in pulmonary arteriolar smooth muscle cells
- This causes membrane depolarization
- Depolarization opens voltage-gated Ca²⁺ channels → Ca²⁺ influx
- Raised intracellular Ca²⁺ → smooth muscle cell contraction and vasoconstriction
Additionally, hypoxia suppresses nitric oxide (NO) synthesis by endothelial cells. Normally NO activates guanylyl cyclase → cGMP → smooth muscle relaxation. In hypoxia, this vasodilator mechanism is blunted, tipping the balance further toward constriction.
Why this becomes pathological in COPD
In COPD, V/Q mismatch is widespread and global - not just in one lung segment. When most alveoli are hypoxic, HPV becomes global rather than local. Instead of a helpful local reflex, it becomes a disease-wide vasoconstriction that raises total PVR and PA pressure throughout the pulmonary circulation. Hypercapnia (which accompanies severe COPD) potentiates HPV, making it even stronger.
Mechanism 2: Vascular Remodeling (Structural - Irreversible)
Chronic hypoxia and cigarette smoke-driven inflammation cause permanent structural changes in pulmonary vessel walls, turning functional (reversible) vasoconstriction into fixed (irreversible) narrowing:
| Layer | Change |
|---|---|
| Intima | Thickening with smooth muscle cell migration and proliferation |
| Media | Smooth muscle hypertrophy; extension of muscle into normally non-muscularized small arteries |
| Adventitia | Increased extracellular matrix deposition |
These structural changes narrow the vessel lumen permanently, raising PVR even during periods when hypoxia is temporarily corrected (e.g., on oxygen therapy). This explains why PH in COPD is only partially reversible with O2.
The vascular remodeling correlates in severity with the degree of airflow obstruction and the intensity of intravascular inflammation - activated CD8+ T lymphocytes are found within pulmonary arterial walls even in smokers with preserved lung function, before overt COPD develops.
Mechanism 3: Endothelial Dysfunction - The Mediator Imbalance
The healthy pulmonary endothelium maintains a balance of vasodilator and vasoconstrictor signals. In COPD this balance is disrupted:
| Mediator | Normal Role | In COPD |
|---|---|---|
| Nitric oxide (NO) | Potent vasodilator + antiproliferative | Reduced production |
| Prostacyclin (PGI2) | Vasodilator + antiproliferative | Reduced |
| Endothelin-1 (ET-1) | Potent vasoconstrictor + promitogenic | Increased |
The net result: a pro-vasoconstriction, pro-proliferative milieu that compounds both acute HPV and chronic remodeling. Endothelial dysfunction has been demonstrated even in patients with mild-to-moderate COPD, meaning this process starts early.
Mechanism 4: Destruction of the Vascular Bed (Anatomical Loss)
Emphysema physically destroys alveolar walls - and with them, the vast capillary network that surrounds each alveolus. This reduces the total cross-sectional area available for blood to flow through.
Think of it like removing lanes from a highway: the same traffic (cardiac output) must now pass through fewer lanes, which increases resistance (pressure rises for the same flow).
Normally, the pulmonary circulation has enormous reserve - it can lose up to 50% of its vascular surface area before resting PA pressure rises, because unused vessels get recruited to compensate. But once emphysematous destruction exceeds this threshold, PA pressure begins to climb and cannot be reversed because the vascular bed is permanently gone.
Mechanism 5: Polycythemia - Increased Blood Viscosity
Chronic hypoxia stimulates the kidneys to produce erythropoietin, driving secondary polycythemia (elevated hematocrit). Thicker, more viscous blood offers greater resistance to flow through the pulmonary vasculature:
Resistance ∝ Viscosity (Poiseuille's Law)
So polycythemia adds a further component to PVR elevation, compounding the vasoconstriction and structural narrowing already present.
Mechanism 6: Mechanical Effects of Hyperinflation
In COPD, gas trapping causes the lungs to be chronically overinflated. The enlarged, distended lung physically compresses alveolar capillaries - particularly extra-alveolar vessels - raising resistance mechanically. This effect is most pronounced during exacerbations when air trapping worsens.
Mechanism 7: Hypercapnia and Acidosis
Chronic CO2 retention (Type 2 respiratory failure) causes respiratory acidosis. Acidosis independently potentiates pulmonary vasoconstriction by:
- Directly increasing vascular smooth muscle tone
- Potentiating the vasoconstrictive response to hypoxia
This creates a vicious cycle: worsening COPD → CO2 retention → acidosis → more pulmonary vasoconstriction → higher PA pressure.
Putting It All Together
Here is how these mechanisms converge step by step as COPD progresses:
Early COPD
↓ V/Q mismatch → local HPV (adaptive, no net PH yet)
↓ Cigarette smoke → endothelial dysfunction, early vascular inflammation
Moderate COPD
↓ Widespread alveolar hypoxia → global HPV → PVR begins to rise
↓ Vascular remodeling progresses → intimal/medial thickening
↓ Emphysema destroys capillary bed → anatomical PVR rise
Severe COPD
↓ Polycythemia (from sustained hypoxia) → hyperviscosity
↓ Hypercapnia and acidosis → potentiate vasoconstriction
↓ Hyperinflation → mechanical compression of vessels
Result: Mean PA Pressure >25 mmHg → Cor Pulmonale
Key Summary Table
| Mechanism | Type | Reversible? | When Dominant |
|---|---|---|---|
| Hypoxic pulmonary vasoconstriction | Functional | Partially (O2 therapy) | Throughout |
| Vascular remodeling | Structural | No | Moderate-severe COPD |
| Endothelial dysfunction (↓NO, ↑ET-1) | Structural/Functional | Partially | Early onset |
| Capillary bed destruction | Anatomical | No | Emphysema-predominant |
| Polycythemia / hyperviscosity | Functional | Yes (treat hypoxia) | Severe hypoxemia |
| Hyperinflation | Mechanical | Partially (bronchodilation) | COPD exacerbations |
| Hypercapnia + acidosis | Functional | Yes (ventilation, O2) | Type 2 respiratory failure |
This is why long-term oxygen therapy is the only treatment that meaningfully lowers PA pressure in COPD - it directly addresses the dominant driver (hypoxic vasoconstriction). But because structural remodeling and anatomical destruction are irreversible, LTOT only stabilizes or mildly reduces PH rather than normalizing it.
Sources: Fuster & Hurst's The Heart 15th Ed. (Ch. 59); Costanzo Physiology 7th Ed.; Fishman's Pulmonary Diseases & Disorders; Murray & Nadel's Textbook of Respiratory Medicine.
This is a shared conversation. Sign in to Orris to start your own chat.