Lipids biochemistry and metabolism
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Let me compile the full answer.
Lipids: Biochemistry and Metabolism
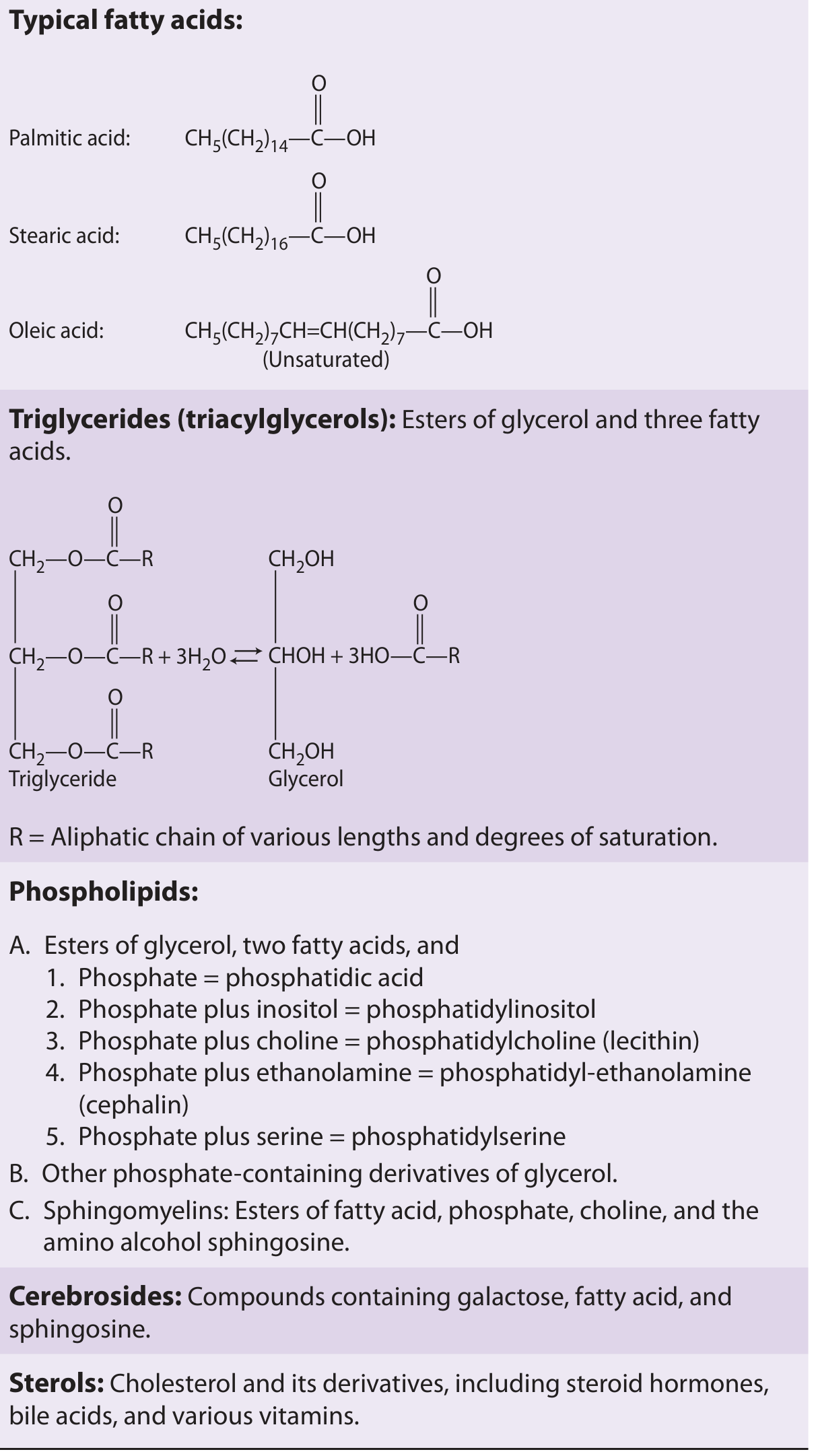
1. Classification of Lipids
Lipids are a chemically diverse group of hydrophobic or amphipathic molecules. The major classes are:
| Class | Structure | Examples |
|---|---|---|
| Fatty acids | Long hydrocarbon chain + carboxyl group | Palmitic (C16:0), Stearic (C18:0), Oleic (C18:1) |
| Triglycerides (triacylglycerols) | Glycerol + 3 fatty acids | Storage fat |
| Phospholipids | Glycerol + 2 FA + phosphate + polar head | Phosphatidylcholine (lecithin), phosphatidylserine |
| Sphingolipids | Sphingosine backbone | Sphingomyelin, cerebrosides |
| Sterols | Steroid nucleus | Cholesterol, steroid hormones, bile acids, vitamin D |
- Saturated fatty acids have no double bonds (e.g., palmitic acid).
- Unsaturated fatty acids have one or more double bonds (e.g., oleic acid has one; linoleic acid has two — an essential fatty acid).
2. Dietary Lipid Digestion & Absorption
Dietary fat is predominantly triglycerides. Key steps:
- Lingual/gastric lipase begins hydrolysis in the stomach.
- Pancreatic lipase (with colipase) cleaves triglycerides to 2-monoglycerides + free fatty acids in the small intestine.
- Bile salts emulsify lipids, forming micelles that allow uptake by enterocytes.
- Inside enterocytes, monoglycerides and fatty acids are resynthesized into triglycerides → packaged into chylomicrons with apolipoprotein B-48.
- Chylomicrons enter the lymphatics (lacteals) → thoracic duct → venous blood at the jugular-subclavian junction.
- Guyton and Hall Textbook of Medical Physiology, p. 842
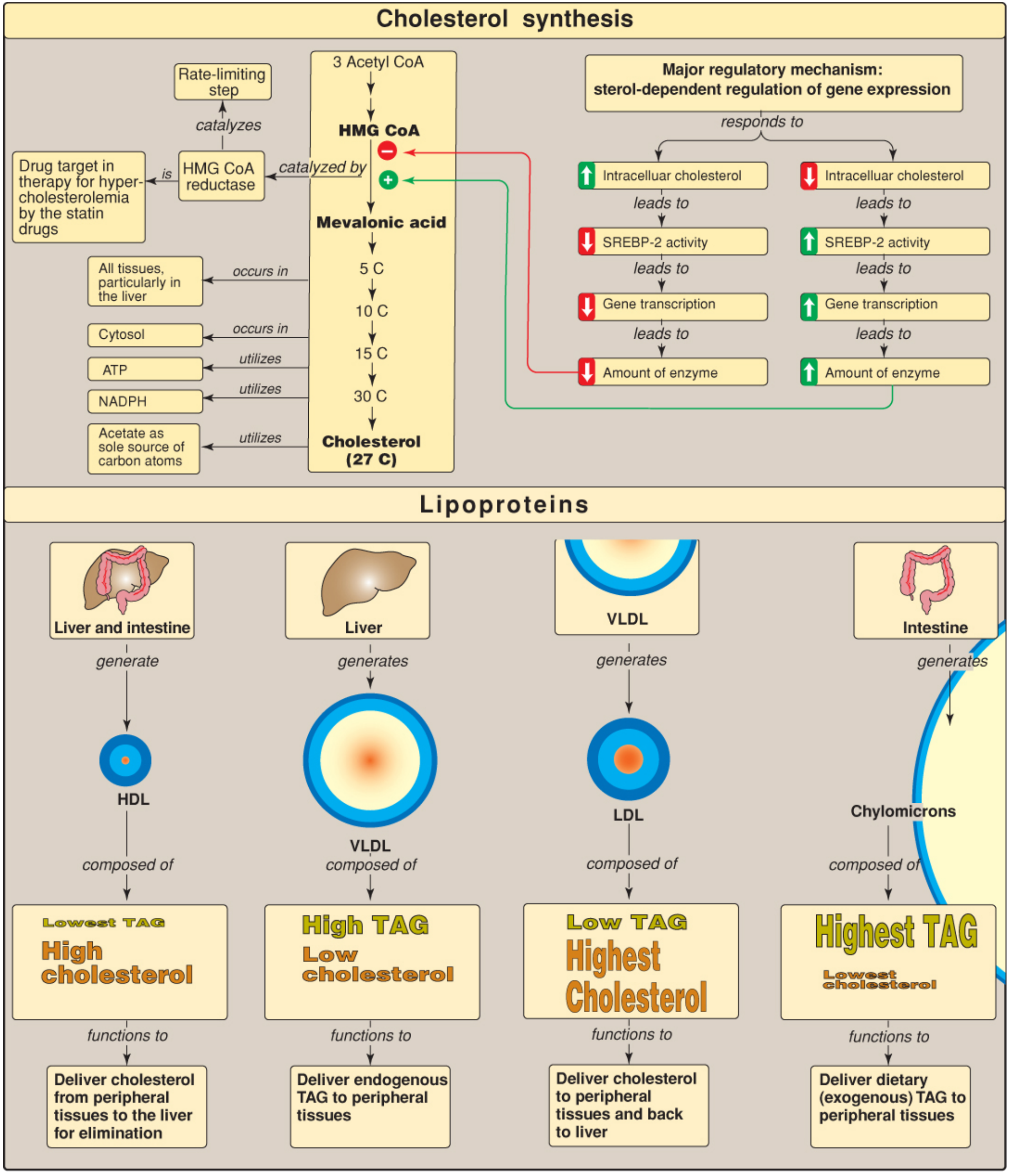
3. Lipid Transport: Lipoproteins
Lipids are insoluble in plasma and are transported as lipoproteins — spherical particles with a hydrophobic core (triglycerides, cholesterol esters) surrounded by an amphipathic shell (phospholipids, free cholesterol, apolipoproteins).
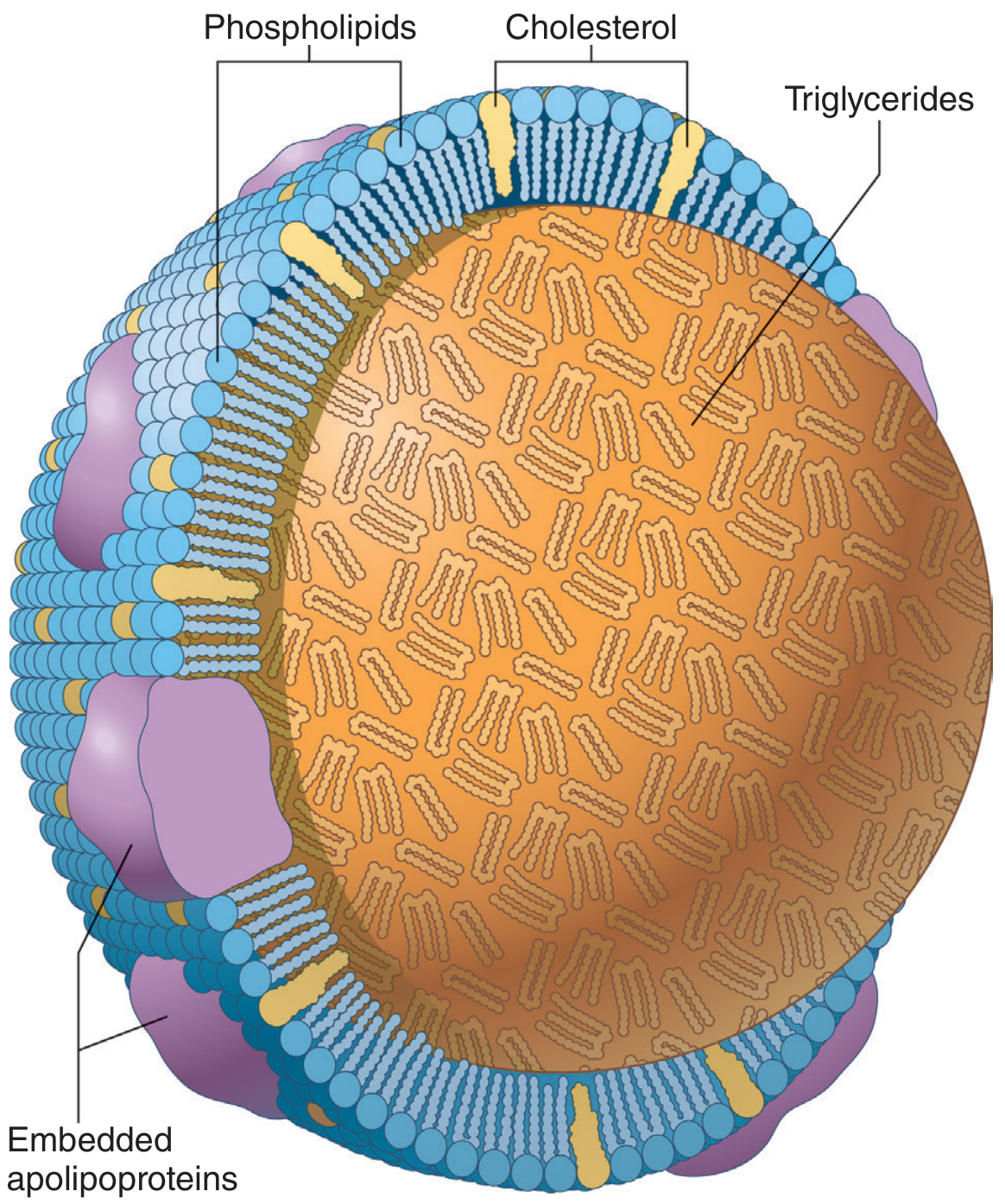
Lipoprotein Classes
| Lipoprotein | Origin | Main Cargo | Key Apolipoprotein | Function |
|---|---|---|---|---|
| Chylomicron | Intestine | Dietary TG (highest) | Apo B-48, Apo C-II, Apo E | Deliver exogenous TG to periphery |
| VLDL | Liver | Endogenous TG (high) | Apo B-100 | Deliver hepatic TG to periphery |
| IDL | From VLDL | TG + cholesterol | Apo B-100, Apo E | Intermediate; cleared by liver or → LDL |
| LDL | From IDL | Cholesterol (highest) | Apo B-100 | Deliver cholesterol to tissues |
| HDL | Liver + intestine | Cholesterol (high protein) | Apo A-I | Reverse cholesterol transport |
- Guyton and Hall Textbook of Medical Physiology, p. 843
- Lippincott's Biochemistry, p. 673
Key Enzymes in Lipoprotein Metabolism
- Lipoprotein lipase (LPL): On capillary endothelium; activated by Apo C-II; hydrolyzes TG in chylomicrons and VLDL → releases fatty acids for storage or energy.
- Hepatic lipase: Converts IDL → LDL.
- LCAT (Lecithin-Cholesterol Acyltransferase): Activated by Apo A-I; esterifies free cholesterol in HDL (core of HDL grows).
- CETP (Cholesterol Ester Transfer Protein): Transfers cholesterol esters from HDL to VLDL/LDL.
Reverse Cholesterol Transport (RCT)
HDL is assembled from lipid-poor Apo A-I secreted by the liver and intestine. Peripheral cells export cholesterol via the ABCA1 transporter → loaded onto HDL → LCAT esterifies it → HDL delivers cholesterol esters to the liver via SR-B1 receptor for elimination as bile acids.
4. Fatty Acid Oxidation (β-Oxidation)
β-Oxidation is the primary pathway for catabolizing fatty acids, occurring in the mitochondrial matrix.
Step 1 — Activation: Fatty acid + CoA → Fatty acyl-CoA (uses ATP; occurs in cytoplasm/outer mitochondrial membrane).
Step 2 — Transport into mitochondria:
- Short- and medium-chain FAs enter freely.
- Long-chain FAs require carnitine shuttle:
- Acyl-CoA + Carnitine → Acylcarnitine (catalyzed by CPT-I on outer membrane)
- Translocase shuttles acylcarnitine across inner membrane
- CPT-II regenerates Acyl-CoA in the matrix
- Carnitine is synthesized from lysine + methionine.
Step 3 — β-Oxidation spiral (repeating 4-step cycle):
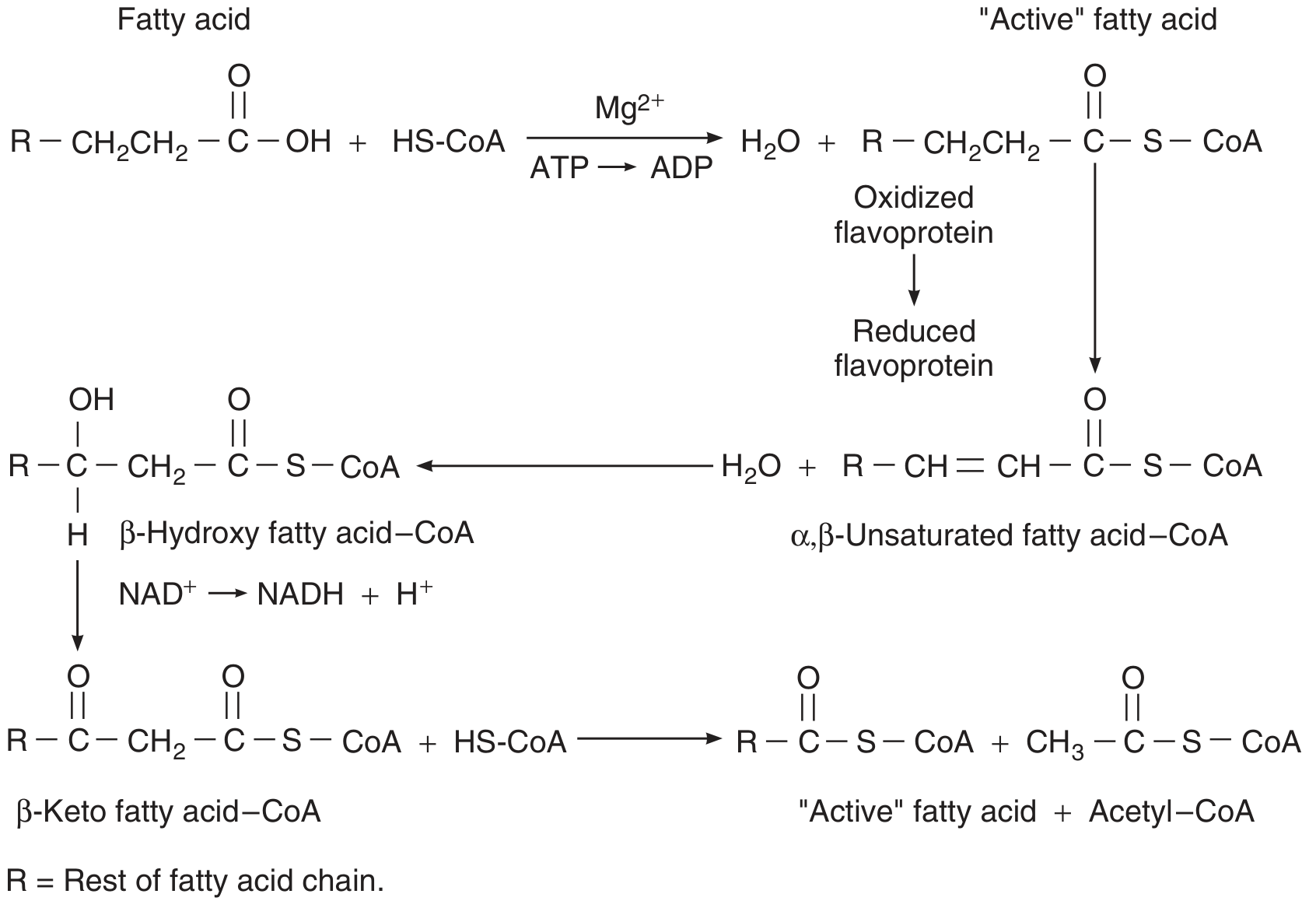
| Step | Reaction | Cofactor |
|---|---|---|
| 1. Oxidation | Acyl-CoA → 2,3-Enoyl-CoA | FAD → FADH₂ |
| 2. Hydration | Enoyl-CoA → 3-Hydroxyacyl-CoA | — |
| 3. Oxidation | 3-Hydroxyacyl-CoA → 3-Ketoacyl-CoA | NAD⁺ → NADH |
| 4. Thiolysis | 3-Ketoacyl-CoA + CoA → Acyl-CoA (–2C) + Acetyl-CoA | — |
Each cycle shortens the chain by 2 carbons, releasing one acetyl-CoA, one FADH₂, and one NADH. Acetyl-CoA enters the TCA cycle.
Energy yield: Catabolism of 1 mol of a 6-carbon fatty acid yields 44 mol ATP vs. 38 mol ATP from glucose — fatty acids are far more energy-dense than carbohydrates.
- Ganong's Review of Medical Physiology, p. 37
5. Ketone Body Formation & Metabolism
When acetyl-CoA production exceeds TCA cycle capacity (starvation, diabetes, high-fat diet), the liver diverts acetyl-CoA to ketone body synthesis:
Pathway (in liver mitochondria):
- 2 Acetyl-CoA → Acetoacetyl-CoA
- Acetoacetyl-CoA + Acetyl-CoA → HMG-CoA (mitochondrial HMG-CoA synthase)
- HMG-CoA → Acetoacetate + Acetyl-CoA (HMG-CoA lyase)
- Acetoacetate → β-Hydroxybutyrate (reversible) or Acetone (irreversible, volatile)
Ketone bodies (acetoacetate, β-hydroxybutyrate, acetone) are exported to peripheral tissues (brain, heart, muscle), where they are converted back to acetyl-CoA → TCA cycle.
Ketoacidosis occurs when ketone body production exceeds peripheral utilization — pH falls, characteristic acetone breath develops. Triggered by: DKA, starvation, alcoholism.
- Guyton and Hall Textbook of Medical Physiology, p. 846
6. Fatty Acid Synthesis (De Novo Lipogenesis)
Fatty acid synthesis is the reverse of β-oxidation in direction but uses a different set of enzymes, location, and cofactors.
| Feature | β-Oxidation | De Novo Synthesis |
|---|---|---|
| Location | Mitochondria | Cytoplasm |
| Carrier | CoA | ACP (Acyl Carrier Protein) |
| Reducing agent | FAD, NAD⁺ (oxidized) | NADPH (consumed) |
| Key enzyme | Thiolase | Fatty Acid Synthase (FAS) |
| Key intermediate | Acetyl-CoA | Malonyl-CoA |
Key steps:
- Acetyl-CoA (mitochondrial) → exported to cytoplasm as citrate via the citrate shuttle
- Acetyl-CoA + CO₂ → Malonyl-CoA (catalyzed by Acetyl-CoA Carboxylase, ACC; requires biotin) — the committed, rate-limiting step
- Fatty Acid Synthase (FAS) elongates the chain by 2 carbons per cycle using malonyl-CoA as donor
- NADPH is supplied by the pentose phosphate pathway (HMP shunt)
- The primary product is Palmitate (C16:0); further elongation/desaturation occurs in the ER
Regulation:
- Insulin activates ACC (stimulates lipogenesis)
- Glucagon/epinephrine inactivate ACC via PKA phosphorylation
- AMPK phosphorylates and inactivates ACC when energy is low
- Malonyl-CoA inhibits CPT-I, preventing simultaneous synthesis and oxidation
7. Cholesterol Biosynthesis
All 27 carbons of cholesterol come from acetyl-CoA. Synthesis occurs primarily in the liver, also in the intestine, adrenal cortex, and gonads. The pathway is cytoplasmic (ER).
4 Stages:
Stage 1: Acetyl-CoA → Mevalonate
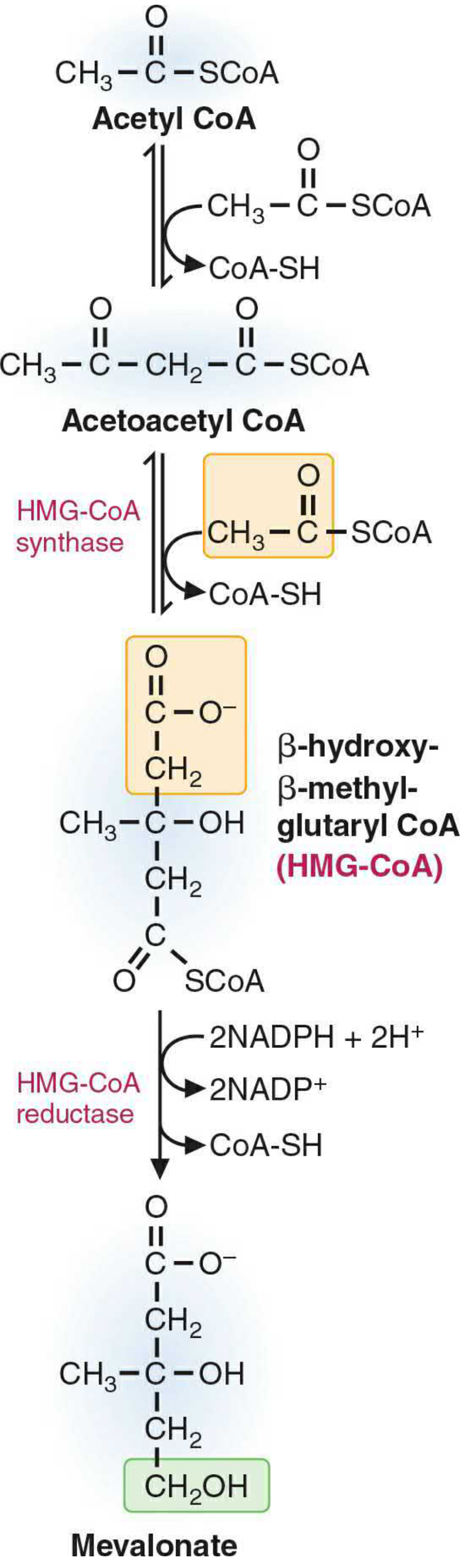
- 2 Acetyl-CoA → Acetoacetyl-CoA → HMG-CoA (cytosolic HMG-CoA synthase)
- HMG-CoA + 2 NADPH → Mevalonate (HMG-CoA reductase) ← rate-limiting step; target of statins
Stage 2: Mevalonate → Activated isoprene (IPP)
- 3 ATP phosphorylate mevalonate → decarboxylation → isopentenyl pyrophosphate (IPP, Δ³-isopentenyl-PP)
Stage 3: IPP → Squalene (C30)
- 6 isoprene units condense (via geranyl-PP, farnesyl-PP) → Squalene (requires NADPH)
Stage 4: Squalene → Cholesterol (C27)
- Squalene cyclizes → lanosterol → multiple steps → cholesterol
Regulation of HMG-CoA Reductase
- SREBP-2 (Sterol Regulatory Element-Binding Protein 2): When intracellular cholesterol is low, SREBP-2 is activated → increases HMG-CoA reductase gene transcription.
- Accelerated protein degradation: High cholesterol promotes enzyme degradation.
- AMPK phosphorylation: Inactivates the enzyme when energy is low.
- Insulin activates; glucagon inhibits.
- Statins: Competitive inhibitors of HMG-CoA reductase → reduce hepatic cholesterol → upregulate LDL receptors → lower plasma LDL.
- Lippincott's Biochemistry, p. 672; Basic Medical Biochemistry 6e
8. Cholesterol Utilization & Elimination
Cholesterol cannot be fully catabolized in humans — its ring structure is intact. It is eliminated by:
- Bile acid synthesis: Rate-limiting step = cholesterol-7α-hydroxylase (inhibited by bile acids — feedback)
- Steroid hormone synthesis: Cholesterol → Pregnenolone (P450scc, rate-limiting) → cortisol, aldosterone, sex hormones
- Vitamin D synthesis: Skin + liver + kidney
- Direct secretion into bile (risk: gallstones / cholelithiasis if bile salts insufficient)
Enterohepatic circulation: >95% of bile acids are reabsorbed in the terminal ileum and returned to the liver. Bile acid sequestrants (cholestyramine) interrupt this cycle.
9. Regulation Summary: Fed vs. Fasted State
| State | Dominant Pathway | Key Hormones |
|---|---|---|
| Fed / Insulin ↑ | FA synthesis, TG storage, cholesterol synthesis | Insulin activates ACC, FAS, HMG-CoA reductase |
| Fasted / Glucagon ↑ | β-Oxidation, ketogenesis, lipolysis | Glucagon/epinephrine activate HSL, inhibit ACC |
| Starvation / DM | Maximal β-oxidation + ketogenesis | Cortisol ↑, glucagon ↑ → FFA release from adipose |
Hormone-sensitive lipase (HSL) in adipocytes is activated by glucagon/epinephrine via cAMP-PKA → hydrolyzes stored TG → releases FFA bound to albumin for transport to other tissues.
10. Clinical Correlations
| Condition | Defect | Consequence |
|---|---|---|
| Familial hypercholesterolemia (FH) | Defective LDL receptor | ↑ LDL-C; premature atherosclerosis |
| Type I hyperlipoproteinemia | LPL or Apo C-II deficiency | Massive chylomicronemia; pancreatitis |
| Type III hyperlipoproteinemia | Apo E2/E2 homozygosity | IDL & chylomicron remnant accumulation |
| Diabetic ketoacidosis | No insulin → no glucose utilization | Massive ketone body production → acidosis |
| MCAD deficiency | Medium-chain acyl-CoA dehydrogenase deficiency | β-Oxidation block → hypoglycemia, hypoketosis |
| Abetalipoproteinemia | No Apo B synthesis | Cannot form chylomicrons/VLDL → fat malabsorption |
| Atherosclerosis | LDL oxidation + macrophage uptake | Foam cells → plaques |
Sources: Guyton and Hall Textbook of Medical Physiology (Chap. 69); Ganong's Review of Medical Physiology, 26e; Lippincott Illustrated Reviews: Biochemistry, 8e; Basic Medical Biochemistry: A Clinical Approach, 6e
Digestion and absorption of lipids
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
I now have comprehensive, detailed content from the textbooks. Let me compile the full answer.
Digestion and Absorption of Lipids
Overview
Lipid digestion and absorption is a multi-step process far more complex than that of carbohydrates or proteins. The fundamental challenge is that dietary lipids are hydrophobic and must be rendered soluble for enzymatic digestion and mucosal uptake in the aqueous environment of the GI tract. Five major phases can be defined:
- Emulsification — mechanical disruption of fat globules
- Intraluminal digestion — enzymatic hydrolysis by lipases
- Micellar solubilization — bile salts package digestion products
- Mucosal uptake — absorption by enterocytes
- Re-esterification & chylomicron assembly — packaging for lymphatic transport
1. Dietary Lipid Composition
90% of dietary fat is triacylglycerols (TAG / triglycerides) — long-chain fatty acyl esters of glycerol. The remainder includes:
- Phospholipids (~5%) — from cell membranes in animal products
- Cholesterol and cholesteryl esters (~5%)
- Fat-soluble vitamins (A, D, E, K)
Lipids are classified by their interaction with water:
- Nonpolar, water-insoluble: TAG, cholesteryl esters, carotene — remain as oil droplets
- Polar amphiphiles: phospholipids, bile acids — form monolayers and micelles
- Medical Physiology (Boron & Boulpaep), p. 1375
2. Emulsification
Effective lipase action requires a large oil-water interface. This is achieved by emulsification — breaking large fat globules into fine droplets.
Mechanical factors:
- Mastication (chewing) and cooking begin the process
- Gastric churning: antral peristalsis against a closed pylorus grinds food into fine particles
- Retrograde propulsion through the contracted pylorus further reduces droplet size
- Intestinal peristalsis mixes luminal contents with pancreatic/biliary secretions
Chemical stabilization of emulsion:
The droplets are coated and stabilized by:
- Biliary phospholipids and cholesterol (amphiphilic — polar heads project into water)
- Denatured proteins and dietary polysaccharides
- Products of early digestion (fatty acids, monoacylglycerols from gastric lipase)
The core of the emulsion particle = TAG + cholesterol esters + other nonpolar lipids.
- Medical Physiology, p. 1377
3. Intraluminal Digestion
A. Lingual Lipase (Minor in Adults)
- Secreted by serous glands at the base of the tongue
- Active in the mouth and continues in the stomach (stable at acidic pH)
- Preferentially cleaves sn-3 position of TAG → fatty acid + 1,2-DAG
- Minor role in adults; more important in neonates (before pancreatic lipase matures)
B. Gastric Lipase
- Secreted by gastric chief cells (stimulated by gastrin)
- 42-kDa glycoprotein; optimum pH 3–6
- ~15% of total fat digestion occurs in the stomach
- Preferentially cleaves the sn-3 position of TAGs; leaves intact 1,2-DAG
- Clinically important in pancreatic insufficiency: compensates for up to 1/3 of fat digestion
- Products (fatty acids) reach the duodenum → stimulate CCK and GIP release
- Medical Physiology, p. 1377–1378
C. Pancreatic Lipase (Major Enzyme)
The key enzyme for fat digestion, secreted by pancreatic acinar cells. Secreted at levels ~1000× more than needed, providing a large reserve capacity.
Requirements for activity:
- Alkaline pH (~6–7; pancreatic HCO₃⁻ neutralizes gastric acid)
- Colipase cofactor (10 kDa; secreted as procolipase, activated by trypsin)
- Ca²⁺, fatty acids, bile salts (in the right concentration)
Mechanism:
- Active only at the oil-water interface of TAG droplets
- Surface emulsifiers (phospholipids, proteins) inhibit pancreatic lipase by blocking the interface
- Bile-salt micelles would also displace lipase — this is reversed by colipase
- Colipase anchors lipase to the lipid interface by penetrating the phospholipid coating
- Crystal studies show lipase has a "lid" over its catalytic cleft; colipase-induced conformational change opens the lid, exposing the active site
- Pancreatic lipase cleaves sn-1 and sn-3 positions of TAG → 2-monoacylglycerol (2-MAG) + 2 free fatty acids (the primary products)
D. Other Pancreatic Esterases
| Enzyme | Substrate | Products |
|---|---|---|
| Phospholipase A2 (PLA2) | Phosphatidylcholine | Lysophosphatidylcholine + fatty acid |
| Cholesterol ester hydrolase | Cholesteryl esters | Free cholesterol + fatty acid |
| Non-specific esterase | Fat-soluble vitamin esters | Free vitamins |
- PLA2 is secreted as a pro-enzyme, activated by trypsin; requires bile salts
- Bile salt–stimulated lipase (in human breast milk) also digests DAGs, MAGs, cholesterol esters, and vitamin esters — important in breast-fed neonates
- Medical Physiology, p. 1378–1380
4. Micellar Solubilization — The Role of Bile Salts
Bile Salt Chemistry
Bile salts (conjugated bile acids) are amphipathic molecules with:
- Hydrophobic steroid backbone (faces lipid)
- Hydrophilic hydroxyl groups and charged conjugated amino acid (glycine or taurine, facing water)
Above the critical micellar concentration (CMC), bile salts spontaneously aggregate into micelles — spherical or cylindrical structures with hydrophobic cores.
From Emulsion Droplet to Mixed Micelle
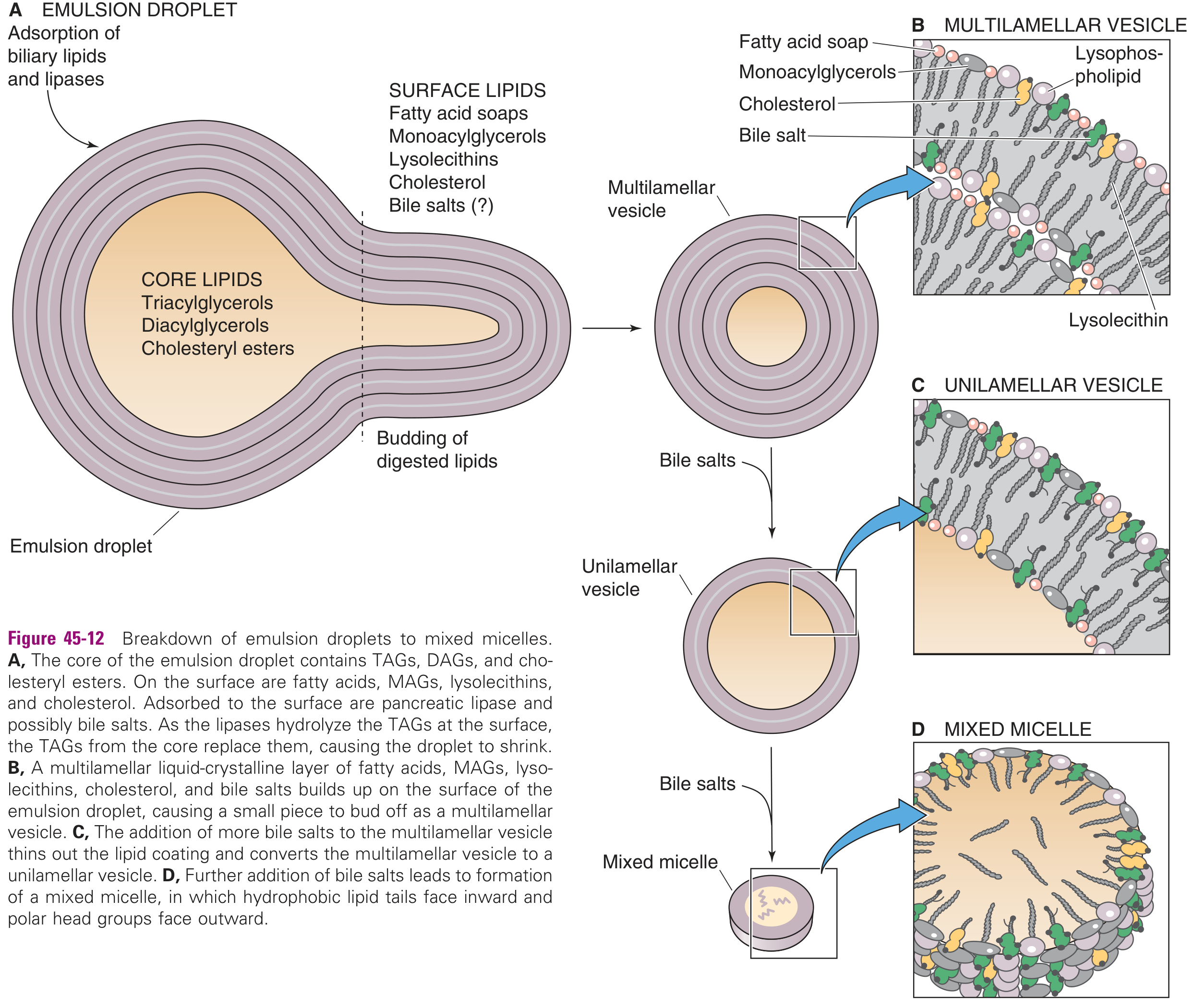
The sequence (as lipases act on emulsion droplets):
- Emulsion droplet (A): Lipases and biliary lipids adsorb to the surface; TAG in core is hydrolyzed
- Multilamellar vesicle (B): Lipolytic products (MAGs, fatty acids, lysophospholipids, cholesterol, bile salts) build up at the surface and bud off as multilayered liquid-crystalline vesicles
- Unilamellar vesicle (C): Bile salts thin the multilamellar coating → single lipid bilayer vesicle
- Mixed micelle (D): Further bile salts completely convert to mixed micelles — hydrophobic tails inward, polar heads outward (~4–8 nm diameter)
Mixed micelles contain: fatty acids + 2-MAG + lysophospholipids + cholesterol + bile salts
Significance: Mixed micelles dramatically increase the aqueous solubility of lipid digestion products, allowing them to diffuse across the unstirred water layer to reach the enterocyte brush border.
When intraluminal bile salt concentrations are low (neonates, obstructive jaundice), absorption can still occur from vesicles, but is less efficient.
- Medical Physiology, p. 1381
5. Mucosal Uptake by Enterocytes
Barriers to Cross
Before entering the enterocyte, lipids must cross:
- Mucous gel layer (95% water, but limits diffusion of large vesicles)
- Unstirred water layer (UWL) — the disequilibrium zone adjacent to the brush border; diffusion is rate-limiting for very lipophilic molecules
- Apical brush-border membrane of enterocytes
How Lipids Enter
Mixed micelles and monomers diffuse from the bulk lumen phase through the mucous gel and UWL to the apical surface. At the brush border:
- The acidic microclimate (pH ~5.5–6.0) at the brush-border surface causes bile salts to dissociate from the micelle (bile salts become protonated at low pH and less micellar)
- Fatty acids and MAGs become protonated and diffuse passively across the apical membrane
Transport mechanisms:
- Long-chain fatty acids (LCFAs) — primarily passive diffusion down concentration gradient; possibly also protein-mediated (FATP4/CD36 transporters on brush border)
- 2-MAG — passive diffusion
- Cholesterol — partially passive, partially via NPC1L1 transporter (target of ezetimibe)
- Short/medium-chain fatty acids (SCFAs/MCFAs) — water-soluble enough to pass directly into the portal blood without re-esterification
Bile salts are NOT absorbed here — they continue to the terminal ileum, where active Na⁺-coupled reabsorption occurs (ASBT transporter), completing enterohepatic circulation.
6. Intracellular Re-esterification and Chylomicron Assembly
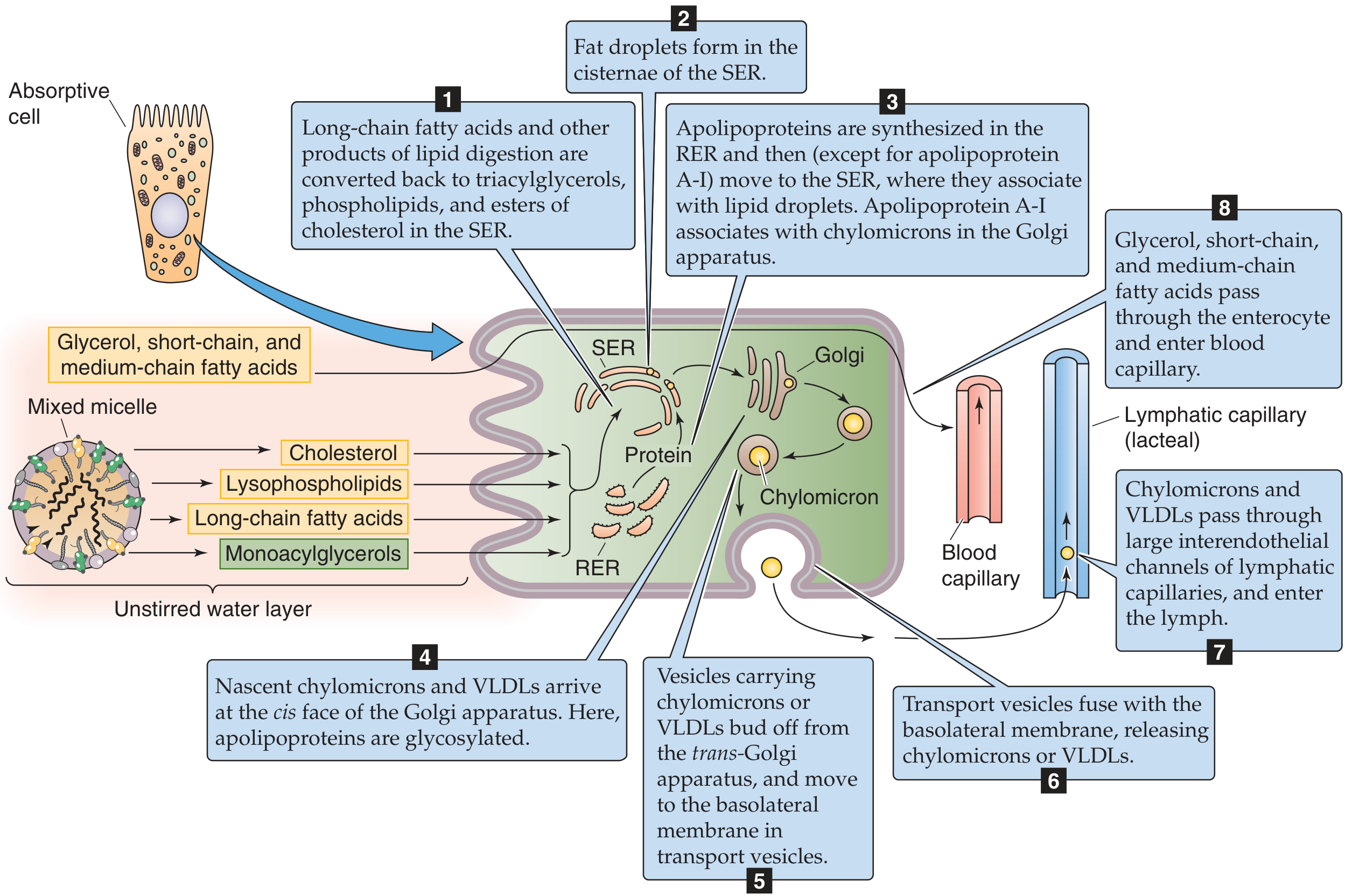
Inside the enterocyte:
Step 1 — Re-esterification (in the Smooth ER):
- LCFAs are activated to Fatty acyl-CoA (by acyl-CoA synthetase)
- Two pathways to reform TAG:
- Monoacylglycerol pathway (dominant during feeding): 2-MAG + 2 acyl-CoA → TAG
- Phosphatidic acid (glycerol-3-phosphate) pathway (fasting): De novo synthesis
- Cholesterol is re-esterified by ACAT (acyl-CoA:cholesterol acyltransferase)
- Lysophospholipids are re-acylated to form phosphatidylcholine (lecithin)
Step 2 — Chylomicron assembly (in the SER and RER):
- Lipid droplets form in the SER cisternae
- Apolipoproteins (especially Apo B-48) are synthesized in the RER → move to SER to associate with lipid droplets
- MTP (Microsomal Triglyceride Transfer Protein) is essential — transfers lipids onto nascent Apo B-48; MTP mutations → abetalipoproteinemia
Step 3 — Maturation and secretion:
- Nascent chylomicrons → Golgi apparatus (for glycosylation of apolipoproteins, addition of Apo A-I)
- Transport vesicles bud from trans-Golgi → move to basolateral membrane
- Vesicles fuse with basolateral membrane → exocytosis of chylomicrons into the lamina propria
Step 4 — Entry into lymph:
- Chylomicrons (~80–500 nm) are too large to enter blood capillary fenestrae
- They pass through larger interendothelial channels into lymphatic capillaries (lacteals)
- Lacteal → cisterna chyli → thoracic duct → left subclavian vein → systemic circulation
Short/medium-chain fatty acids (C6–C12) bypass this pathway — they are water-soluble, do not require re-esterification, and pass directly into portal blood → liver. This is why MCT (medium-chain triglyceride) formulas are useful in fat malabsorption syndromes.
- Medical Physiology, p. 1383–1385
7. Cholesterol and Phospholipid Absorption (Summary)
| Lipid | Digestion | Absorption | Transport |
|---|---|---|---|
| Cholesterol esters | Cholesterol ester hydrolase → free cholesterol | Via micelles → NPC1L1 | Re-esterified (ACAT) → chylomicron |
| Free cholesterol | None needed | As above | As above |
| Phospholipids | PLA2 → lyso-PC + FA | Via micelles → passive | Re-acylated → chylomicron or VLDL |
| Fat-soluble vitamins | Ester hydrolases | Via micelles (require bile salts) | Chylomicrons |
8. Enterohepatic Circulation of Bile Salts
- Bile salts are efficiently reabsorbed in the terminal ileum (>95%) via the ASBT (apical sodium-dependent bile acid transporter)
- Returned to liver via portal vein → re-secreted into bile
- Normal bile salt pool: ~3–5 g; recirculates 6–10 times/day
- Interruption (ileal resection, cholestyramine) → increased synthesis from cholesterol → lowers cholesterol; also → fat malabsorption if pool is too depleted
- Costanzo Physiology 7e, p. 387
9. Clinical Consequences of Impaired Lipid Digestion/Absorption
All steps are necessary — failure at any point → steatorrhea (fat in stool):
| Cause | Mechanism | Examples |
|---|---|---|
| Pancreatic insufficiency | No lipase/colipase/PLA2 | Chronic pancreatitis, cystic fibrosis |
| Low duodenal pH | Lipase inactivated below pH 5 | Zollinger-Ellison syndrome |
| Bile salt deficiency | No micelle formation | Ileal resection, obstructive jaundice, bacterial overgrowth (bile salt deconjugation) |
| Mucosal disease | Reduced absorptive surface | Celiac disease (villous atrophy), Crohn's, short bowel syndrome |
| MTP deficiency | No chylomicron assembly | Abetalipoproteinemia (no Apo B-48; fat malabsorption + acanthocytosis + vitamin E deficiency) |
| Lymphatic obstruction | Chylomicrons cannot exit | Intestinal lymphangiectasia (protein-losing enteropathy + steatorrhea) |
Zollinger-Ellison syndrome illustrates two mechanisms simultaneously: excess gastric acid delivered to duodenum (1) inactivates pancreatic lipase and (2) damages intestinal mucosa — both → steatorrhea. Costanzo Physiology 7e, p. 387–388
Summary Diagram of the Complete Process
Dietary TAG (>90%)
│
▼
EMULSIFICATION (stomach churning + biliary phospholipids)
│
▼
GASTRIC LIPASE → ~15% TAG hydrolyzed → 1,2-DAG + FA
│ (CCK released in duodenum)
▼
PANCREATIC LIPASE + COLIPASE → 2-MAG + 2 FA ← PLA2 (phospholipids → lyso-PC)
← Cholesterol ester hydrolase
│
▼
BILE SALTS form MIXED MICELLES (fatty acids, 2-MAG, lyso-PC, cholesterol)
│
▼ (diffuse through unstirred water layer)
ENTEROCYTE BRUSH BORDER UPTAKE
LCFAs + 2-MAG → passive diffusion / FATP4/CD36
Cholesterol → NPC1L1
SCFAs/MCFAs → portal blood directly
│
▼
RE-ESTERIFICATION in SER (TAG, phospholipids, cholesterol esters)
│
▼
CHYLOMICRON ASSEMBLY (Apo B-48, MTP) in SER/RER → Golgi → vesicles
│
▼
EXOCYTOSIS at basolateral membrane → LACTEALS → thoracic duct → blood
│
▼
Bile salts → terminal ileum ASBT reabsorption → portal vein → ENTEROHEPATIC CIRCULATION
Sources: Medical Physiology (Boron & Boulpaep), Chap. 45, pp. 1375–1385; Costanzo Physiology, 7e, Chap. 8, pp. 386–388; Yamada's Textbook of Gastroenterology, 7e, p. 100
Source: Please from Vasudevan
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
Create a summary PDF on lipid digestion & absorption
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
Urinary bladder
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
This is a shared conversation. Sign in to Orris to start your own chat.