Cardiomyopathy
Reading File
Finding Sources
Searching PubMed
"cardiomyopathy"[MeSH Terms] AND ("classification" OR "management")
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
I now have all the content I need for a comprehensive response. Let me compose the full answer.
Cardiomyopathy
Cardiomyopathies are intrinsic diseases of cardiac muscle - distinct from myocardial dysfunction caused by coronary artery disease, hypertension, valvular disease, or congenital defects. They are classified into three major functional patterns based on pathophysiology.
Classification Overview
| Type | LV Ejection Fraction | Dysfunction | Prevalence |
|---|---|---|---|
| Dilated (DCM) | <40% | Systolic (contractile) | ~90% of cardiomyopathies |
| Hypertrophic (HCM) | 50-80% | Diastolic (relaxation) | ~1 in 500 |
| Restrictive | Near-normal | Diastolic (compliance) | Least common |
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Table 12.5
1. Dilated Cardiomyopathy (DCM)
Definition & Pathophysiology
DCM is characterized by progressive cardiac dilation and contractile (systolic) dysfunction, usually with concomitant hypertrophy. All four chambers dilate, but LV failure dominates. The heart appears flabby and hypocontracting.
Causes / Pathogenesis
Several pathways converge on the same end-stage phenotype:
- Genetic (20-50% of cases): Over 50 genes implicated, predominantly autosomal dominant inheritance. Loss-of-function mutations in cytoskeletal, sarcolemmal, or nuclear envelope proteins. The most common are titin (TTN) truncation mutations, accounting for 10-20% of all DCM. Other genes: β-myosin heavy chain, cardiac troponin T, desmin, lamin A/C, dystrophin (X-linked; associated with Duchenne/Becker muscular dystrophy).
- Viral myocarditis: Coxsackievirus B, adenovirus, parvovirus B19, HHV-6. Sequential biopsies have documented progression from myocarditis to DCM.
- Alcohol/toxins: Ethanol and its metabolite acetaldehyde are directly cardiotoxic. Chronic alcohol use may also cause thiamine deficiency (beriberi heart disease). Chemotherapy agents (notably doxorubicin/anthracyclines) are important toxic causes.
- Peripartum cardiomyopathy: Occurs in the last trimester or up to 5 months postpartum; likely multifactorial (genetic susceptibility, volume overload, nutritional factors).
- Other: Hemochromatosis, sarcoidosis, chronic anemia, cobalt toxicity, idiopathic.
Morphology
- All four chambers dilated; heart is heavy (up to 900 g)
- Mural thrombi common, especially at the LV apex (risk of systemic embolism)
- Histology: myocyte hypertrophy, nuclear enlargement, interstitial fibrosis - all nonspecific
Gross and histological appearance of DCM:
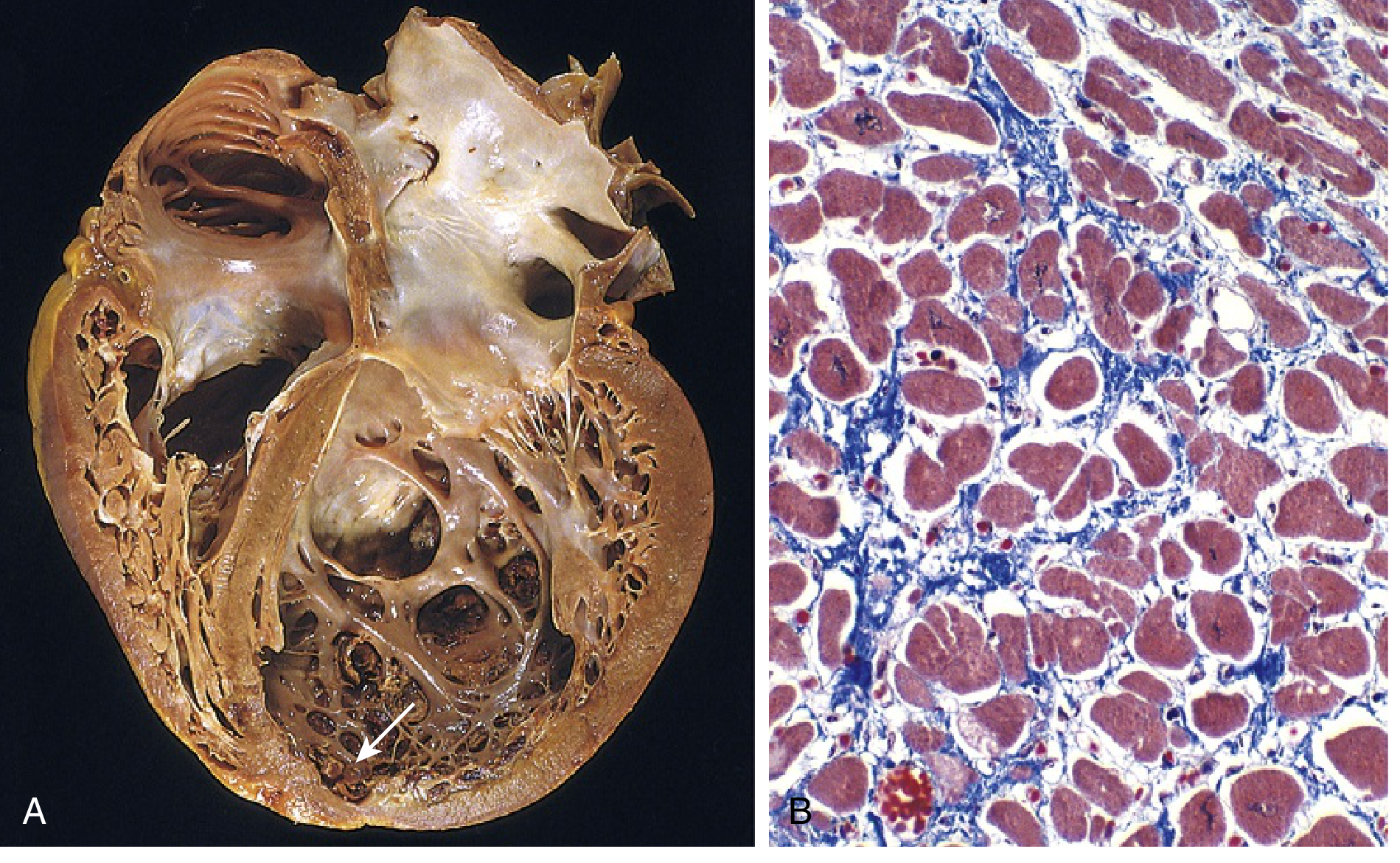
(A) Four-chamber dilation; mural thrombus at LV apex (arrow). (B) Myocyte hypertrophy and interstitial fibrosis (collagen = blue, Masson trichrome) - Robbins Basic Pathology, Fig. 9.25
Clinical Features
- Symptoms of progressive heart failure: dyspnea, fatigue, peripheral edema
- Dilated ventricle promotes dispersion of ventricular depolarization/repolarization - substrate for ventricular tachyarrhythmias and sudden cardiac death
- Mural thrombi - embolic stroke risk
- NYHA class II-III patients: higher risk of sudden cardiac death than pump failure; class IV: more likely to die of pump failure
2. Hypertrophic Cardiomyopathy (HCM)
Definition & Pathophysiology
HCM is characterized by massive myocardial hypertrophy without ventricular dilation, defective diastolic filling, and - in approximately one-third of cases - ventricular outflow obstruction (obstructive HCM). Systolic function is preserved (EF 50-80%), but diastolic dysfunction dominates. Prevalence is approximately 1 in 500 - making it the most common inherited cardiac disorder.
Pathogenesis
- Almost all cases are autosomal dominant, gain-of-function mutations in sarcomeric proteins
- More than 400 mutations in at least 9 genes identified
- Most commonly affected genes: β-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), and cardiac troponin T (TNNT2) - together accounting for 70-80% of cases
- These are gain-of-function mutations causing myofilament hypercontractility, increased energy consumption, and net negative energy balance
- Note: some of the same genes (e.g., β-myosin) carry loss-of-function mutations in DCM
Morphology
- Massive myocardial hypertrophy; heart weight often 600-1000 g
- 90% of cases: Disproportionate thickening of the ventricular septum relative to the LV free wall - "asymmetric septal hypertrophy"
- Remaining 10%: concentric hypertrophy
- The thickened septum can bulge into the LV outflow tract
- Histology (pathognomonic): myocyte disarray - haphazardly oriented, interlocking hypertrophic myocytes, with interstitial and replacement fibrosis
Outflow Obstruction (Obstructive HCM)
- The hypertrophied septum narrows the LV outflow tract
- Dynamic obstruction created by systolic anterior motion (SAM) of the anterior mitral leaflet
- SAM causes both outflow obstruction AND mitral regurgitation
- Obstruction is dynamic: worsens with decreased preload (dehydration, Valsalva, standing), decreased afterload, or increased contractility (exercise, inotropes)
Clinical Features
- Dyspnea, angina, syncope (classic triad)
- Harsh systolic ejection murmur along the left sternal border (increases with Valsalva, decreases with squatting)
- Most common cardiovascular cause of sudden cardiac death in young athletes (accounts for ~1/3 of such events)
- Ventricular tachyarrhythmias arise from the substrate of myocyte disarray and fibrosis
- Atrial fibrillation is common; preferred rate/rhythm control: disopyramide + beta-blocker, or verapamil/diltiazem; amiodarone if needed
ICD Indications in HCM
ICD is recommended for patients with:
- Prior cardiac arrest or VF
- First-degree relative with sudden cardiac death
- Unexplained syncope
- LV wall thickness ≥ 30 mm
- Abnormal BP response to exercise with other risk factors
- Hemodynamically significant or non-sustained VT
- High-risk children with unexplained syncope, massive LVH, or family history of SCD
- Tintinalli's Emergency Medicine, p. 95; Braunwald's Heart Disease
3. Restrictive Cardiomyopathy
Definition & Pathophysiology
Restrictive cardiomyopathy is characterized by a decrease in ventricular compliance, resulting in impaired ventricular filling during diastole. The wall is stiff - the ventricle cannot relax and fill normally. Systolic function is typically preserved; EF may be near-normal. Atria dilate due to high filling pressures.
Morphology
- Ventricles approximately normal in size; cavities not dilated; myocardium is firm
- Both atria markedly dilated (due to high filling pressures)
- Microscopy: variable interstitial fibrosis; etiology-specific features on biopsy
Major Causes
1. Cardiac Amyloidosis
- Extracellular deposition of proteins with β-pleated sheet conformation
- Can occur in systemic amyloidosis (e.g., AL amyloid from multiple myeloma/plasma cell dyscrasias) or isolated cardiac form (ATTR - transthyretin amyloidosis)
- ATTR: deposition of normal or mutant transthyretin in older adults
- A specific TTR mutation (Val122Ile) is found in ~4% of African Americans, increasing cardiac amyloidosis risk >4-fold
- AL amyloid light chains are also directly cardiotoxic beyond just mechanical deposition
2. Endomyocardial Fibrosis
- Most common worldwide form of restrictive cardiomyopathy
- Children and young adults in Africa and other tropical regions
- Diffuse fibrosis of ventricular endocardium and subendocardium, involving tricuspid and mitral valves
- Associated with helminthic infections and nutritional deficiencies
3. Loeffler Endomyocarditis
- No geographic predilection
- Peripheral hypereosinophilia + eosinophilic tissue infiltrates
- Eosinophil major basic protein causes endocardial/myocardial necrosis, then scarring, mural thrombus formation, and thrombus organization
- Endocardial fibrosis with large mural thrombi
Other causes: Radiation fibrosis, sarcoidosis, mucopolysaccharide and sphingolipid storage diseases (Fabry, Gaucher, Hurler)
4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
ARVC is an autosomal dominant disorder with variable penetrance, classically manifesting with:
- Right-sided heart failure
- Ventricular arrhythmias of RV origin (VT with left bundle branch block morphology)
- Syncope and sudden cardiac death (especially in young athletes)
Pathogenesis
Most causal mutations involve genes encoding desmosomal junctional proteins at the intercalated disk - e.g., plakoglobin, plakophilin-2, desmoplakin, desmoglein - or proteins interacting with the desmosome (desmin). Disrupted cell-cell junctions lead to myocyte death and replacement by fat and fibrosis.
- Naxos syndrome: ARVC + palmoplantar keratoderma (skin hyperkeratosis) - associated with plakoglobin mutations
Morphology
- Right ventricular wall severely attenuated by myocyte loss
- Near-transmural replacement of RV free wall by fat and fibrosis
- LV involvement possible but less prominent
- Mononuclear inflammation may surround degenerating myocytes
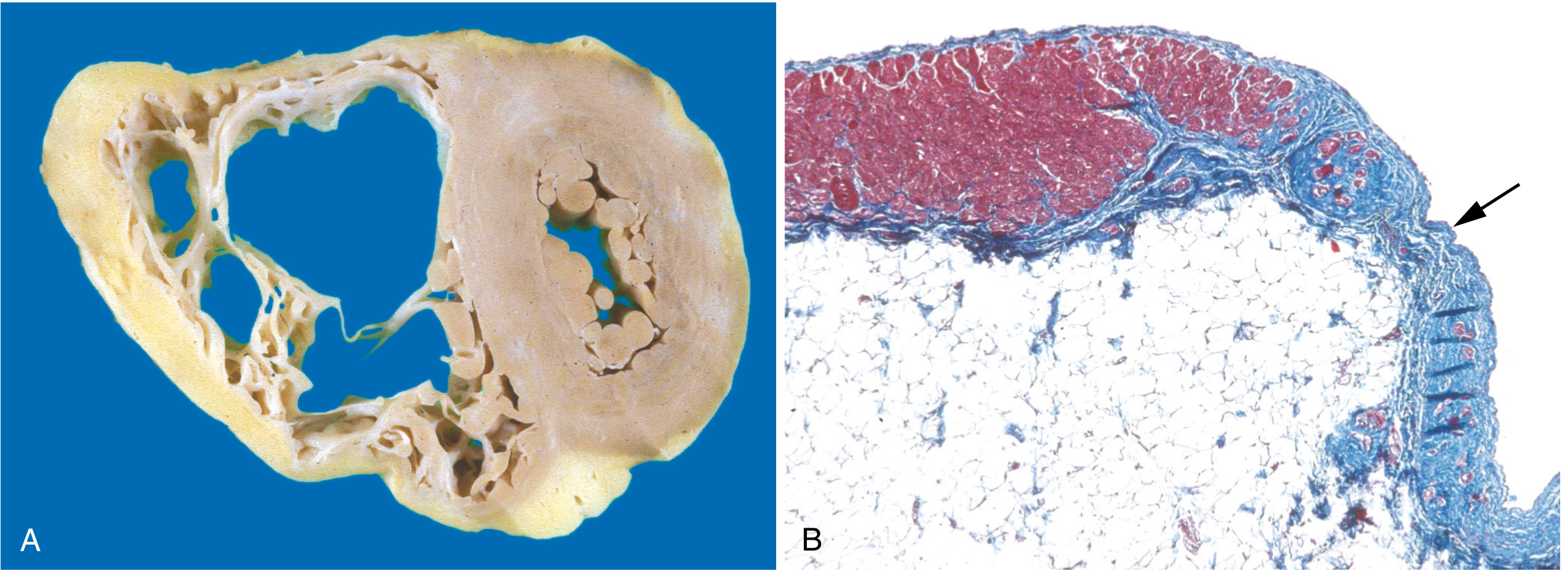
(A) Marked RV dilation; near-transmural RV free wall replacement by fat and fibrosis. (B) Masson trichrome: myocardium (red) replaced by fibrosis (blue, arrow) and fat. - Robbins, Cotran & Kumar Pathologic Basis of Disease, Fig. 12.31
ECG Features
ECG typically shows: epsilon wave (small positive deflection after QRS in right precordial leads), T-wave inversions in V1-V3, right bundle branch block morphology VT.
Summary Comparison Table
| Feature | DCM | HCM | Restrictive | ARVC |
|---|---|---|---|---|
| Ventricular size | Dilated | Normal/small cavity | Normal/small cavity | RV dilated |
| Wall thickness | Thin | Thick | Normal | RV thinned |
| EF | Reduced (<40%) | Normal-high (50-80%) | Normal | Variable |
| Dysfunction | Systolic | Diastolic | Diastolic | Arrhythmia/RHF |
| Key genetics | Titin (TTN), dystrophin, lamin A/C | β-myosin, MYBPC3, TnT | TTR mutations (amyloid) | Desmosomal proteins |
| Mutation type | Loss-of-function | Gain-of-function | - | Desmosome disruption |
| SCD risk | Moderate | High (young athletes) | Lower | High (young athletes) |
| Key finding | Mural thrombi | Myocyte disarray, SAM | Bilateral atrial dilation | Epsilon wave on ECG |
Key Points for Exams
- DCM: titin (TTN) is the most common single gene mutated; loss-of-function sarcomere mutations
- HCM: gain-of-function sarcomere mutations; asymmetric septal hypertrophy in 90%; myocyte disarray is pathognomonic; #1 cause of SCD in young athletes
- HCM murmur: increases with Valsalva (decreased preload), decreases with squatting (increased preload)
- Restrictive: both atria dilated; biventricular filling pressures elevated; amyloid is the key cause in the Western world
- ARVC: desmosomal protein mutations; fatty replacement of RV; epsilon wave; VT with LBBB morphology; Naxos syndrome (+ palmoplantar keratoderma)
- Peripartum cardiomyopathy: a form of DCM; up to 5 months postpartum; generally reversible in ~50% of cases
Sources: Robbins & Kumar Basic Pathology, 10e; Robbins, Cotran & Kumar Pathologic Basis of Disease, 10e; Goldman-Cecil Medicine; Tintinalli's Emergency Medicine, 9e; Braunwald's Heart Disease, 12e; Washington Manual of Medical Therapeutics
This is a shared conversation. Sign in to Orris to start your own chat.