
IMG_20260621_103621_330.jpg
Explain diabetes Mellitus
Reading File
Finding Sources
Searching PubMed
"diabetes mellitus"[MeSH Terms] AND "pathogenesis"[MeSH Terms]
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Here is a comprehensive explanation of Diabetes Mellitus based on Robbins, Cotran & Kumar - Pathologic Basis of Disease (the exact textbook shown in your image).
Diabetes Mellitus
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, Chapter 24: The Endocrine System
Definition
Diabetes mellitus is a group of metabolic disorders sharing the common feature of hyperglycemia caused by defects in insulin secretion, insulin action, or - most commonly - both. Chronic hyperglycemia leads to secondary damage in multiple organ systems, especially the kidneys, eyes, nerves, and blood vessels.
Epidemiology
- Affects >30 million people (>11% of the US population)
- ~1.9 million have Type 1; the vast majority (~90-95%) have Type 2
- ~96 million US adults have prediabetes
- WHO estimates 422 million people with diabetes worldwide
- 7th leading cause of death in the USA
- Total yearly cost in the US: ~$327 billion
Diagnosis (ADA/WHO Criteria)
Normal blood glucose: 70-120 mg/dL
Diabetes is diagnosed by any one of the following (confirmed on a separate day, except random glucose with symptoms):
| Test | Diabetes | Prediabetes |
|---|---|---|
| Fasting plasma glucose | ≥126 mg/dL | 100-125 mg/dL |
| Random plasma glucose | ≥200 mg/dL (with symptoms) | - |
| 2-hr glucose (OGTT, 75g) | ≥200 mg/dL | 140-199 mg/dL |
| HbA1c | ≥6.5% | 5.7-6.4% |
Classification
Type 1 Diabetes (T1D) - ~5-10%
Autoimmune destruction of pancreatic β-cells causing absolute insulin deficiency.
- Most common in patients <20 years, but can occur at any age
- Requires insulin for survival
- Risk of diabetic ketoacidosis (DKA)
- LADA (Latent Autoimmune Diabetes in Adults) is a slowly progressive adult form
Type 2 Diabetes (T2D) - ~90-95%
Combination of peripheral insulin resistance + relative insulin deficiency (inadequate β-cell compensatory response).
- Most individuals are overweight/obese
- Prevalence rising sharply in children and adolescents
Other Forms
- Monogenic diabetes: MODY (Maturity-Onset Diabetes of the Young) - genetic defects in β-cell function or insulin action
- Gestational diabetes: glucose intolerance first recognized during pregnancy
- Secondary causes: pancreatitis, Cushing syndrome, acromegaly, drug-induced
Glucose Homeostasis (Normal)
Glucose is tightly regulated by three processes:
- Hepatic glucose production (gluconeogenesis + glycogenolysis)
- Peripheral glucose uptake (mainly skeletal muscle)
- Insulin/glucagon balance
- Fasting: low insulin, high glucagon → hepatic glucose release prevents hypoglycemia
- Post-meal: insulin rises, glucagon falls → glucose uptake in muscle and fat
Insulin secretion from β-cells: triggered by rising blood glucose → glucose enters β-cell via GLUT2 → metabolized → ↑ATP/ADP ratio → closes K⁺-ATP channels → membrane depolarization → Ca²⁺ influx → insulin granule exocytosis (first-phase rapid, second-phase sustained)
Pathogenesis of Type 1 Diabetes
T1D is an autoimmune disease - immune effector cells attack endogenous β-cell antigens.
Genetic Susceptibility
- HLA gene cluster contributes ~50% of genetic risk
- 90-95% of European patients carry HLA-DR3 or HLA-DR4 (vs ~40% of normal subjects)
- 40-50% of T1D patients are DR3/DR4 compound heterozygotes (vs 5% of normal subjects)
- DR3 or DR4 + DQ8 haplotype = highest inherited risk
- Other susceptibility genes: CTLA4, PTPN22, STAT3, AIRE mutations
Environmental Factors
- Viral infections may trigger islet autoimmunity through molecular mimicry (viral epitopes cross-react with islet antigens)
- Diet, gut microbiome, and hygiene hypothesis also implicated
Mechanisms of β-Cell Destruction
- CD8⁺ cytotoxic T cells directly kill β-cells
- CD4⁺ Th1 cells activate macrophages that produce cytokines damaging β-cells
- Autoantibodies (anti-insulin, anti-GAD65, anti-IA-2) are markers of T1D but not primary effectors
- Progressive loss of β-cell mass over years before clinical presentation
Pathogenesis of Type 2 Diabetes
Two central defects:
1. Insulin Resistance
Tissues (especially skeletal muscle, liver, adipose) fail to respond normally to insulin.
Key contributor - Obesity:
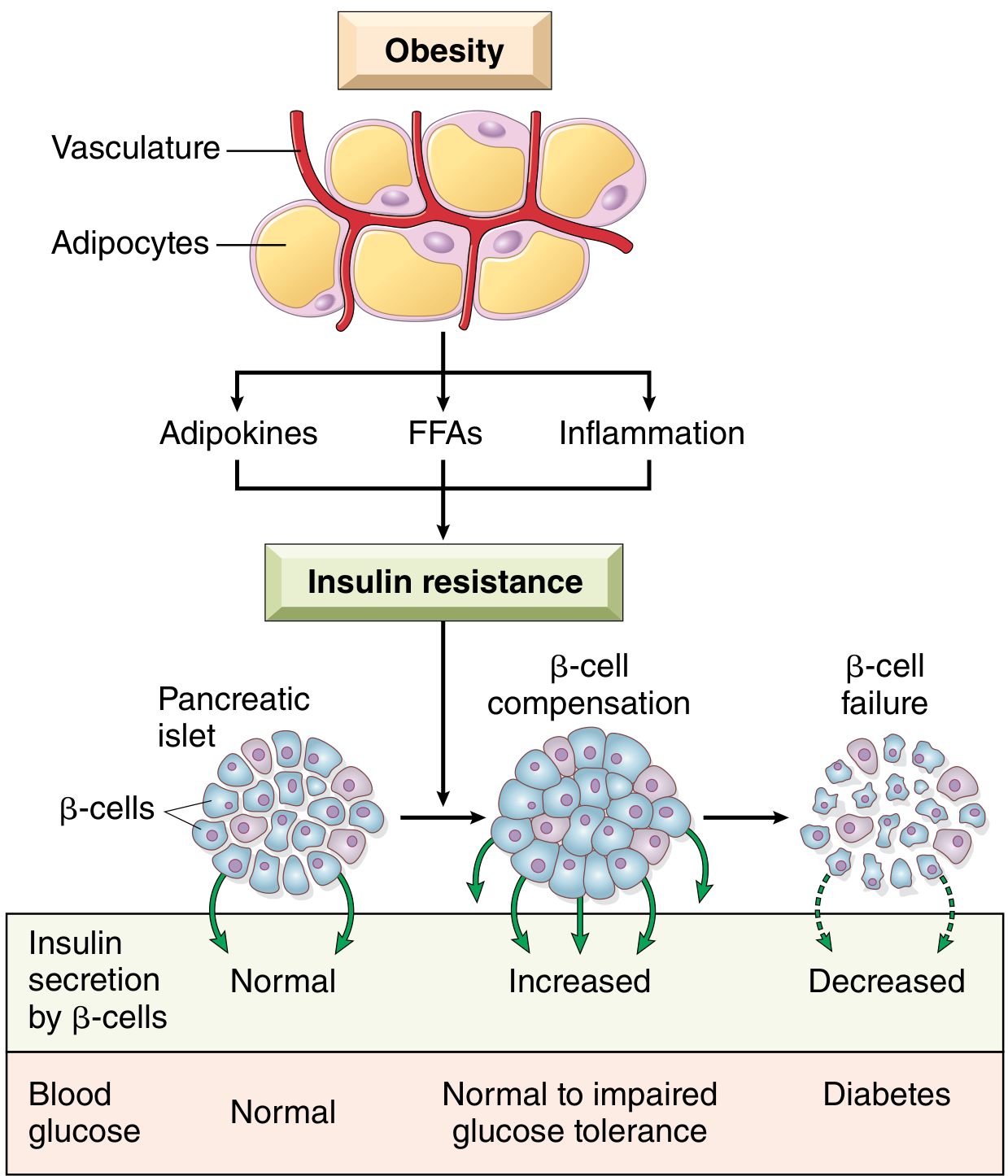
- Free fatty acids (FFAs): Central adipose tissue is highly lipolytic; excess FFAs accumulate toxic lipid intermediates (DAG, ceramides) that impair insulin receptor signaling and activate inflammatory pathways. In the liver, attenuated insulin signaling unleashes phosphoenolpyruvate carboxykinase, driving excess gluconeogenesis.
- Adipokines: Adipose tissue secretes hormones. Adiponectin (which improves insulin sensitivity) is reduced in obesity, worsening resistance.
- Inflammation: Excess FFAs and glucose activate the inflammasome in macrophages and β-cells → IL-1β secretion → proinflammatory cytokine cascade → insulin resistance at peripheral tissues.
2. β-Cell Failure
Initially, β-cells compensate by secreting more insulin (hyperinsulinemia). Over time, sustained demands lead to:
- β-cell exhaustion and reduced mass (partly from amyloid deposition - islet amyloid polypeptide/IAPP)
- Glucotoxicity and lipotoxicity accelerate β-cell dysfunction
- Eventually, insulin secretion is inadequate → frank hyperglycemia
Mechanisms of Vascular Complications (Common to T1D & T2D)
Three major biochemical pathways drive end-organ damage:
1. Advanced Glycation End-products (AGEs)
- Glucose non-enzymatically glycates proteins and lipids → AGEs
- AGEs bind RAGE receptors on endothelial cells, smooth muscle, macrophages → release of cytokines (TGF-β, VEGF), ROS, and procoagulant factors → basement membrane thickening, vascular injury
2. Protein Kinase C (PKC) Activation
- Hyperglycemia → excess diacylglycerol (DAG) synthesis → PKC activation → overproduction of VEGF (retinopathy), TGF-β (glomerulosclerosis), and PAI-1 (thrombosis)
3. Polyol Pathway & Oxidative Stress
- In insulin-independent tissues (nerves, lens, kidney, vessels): excess glucose → aldose reductase converts glucose to sorbitol (polyol pathway)
- This depletes NADPH → impairs glutathione regeneration → ↑ oxidative stress
- Sorbitol accumulation in the lens contributes to cataracts
Long-Term Complications
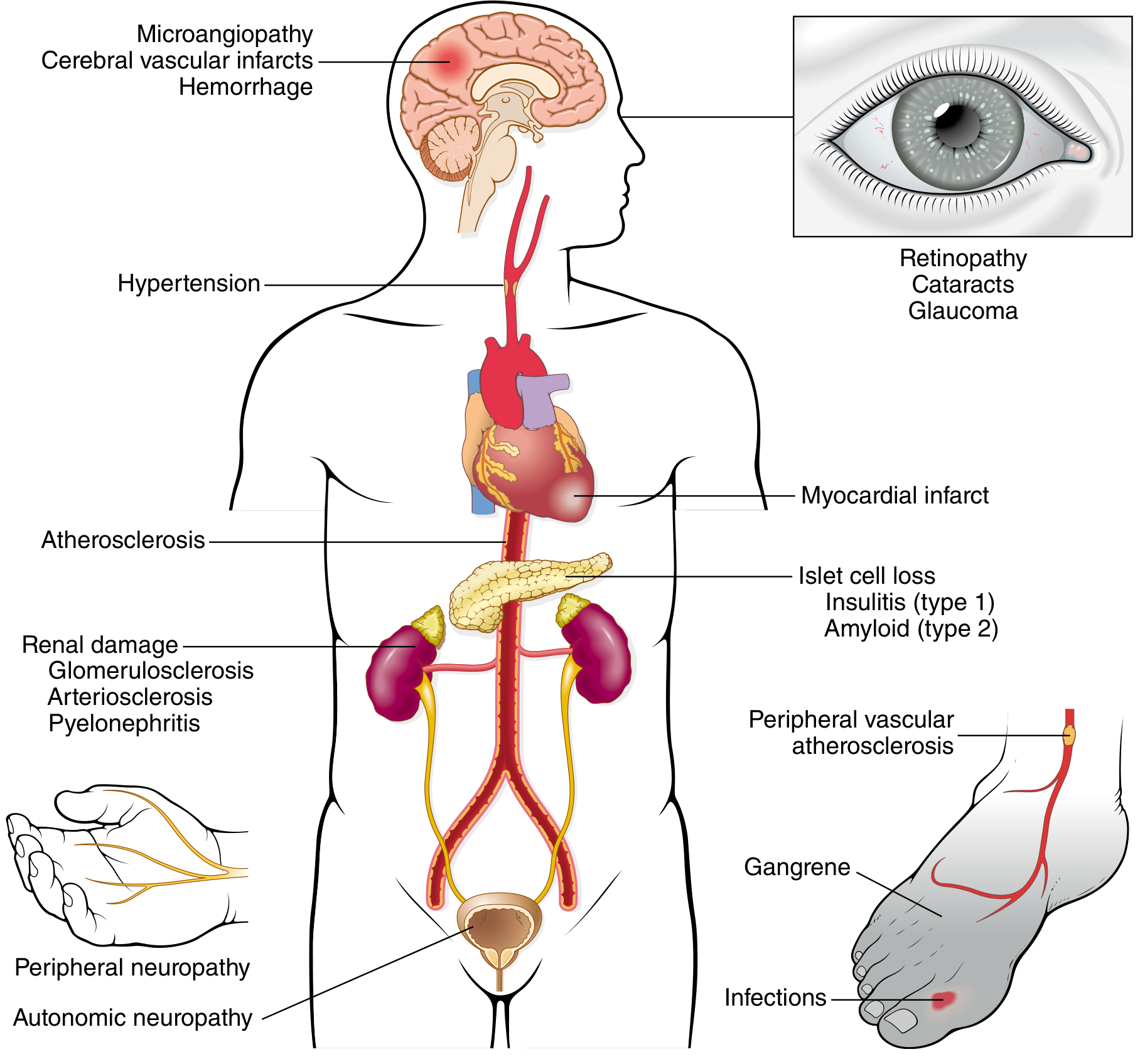
Macrovascular Disease
- Most common cause of death in long-standing diabetes
- 2-4× higher risk of coronary artery disease
- 4× higher risk of dying from cardiovascular complications
- Risk elevated even at the prediabetes stage
- ~75% of T2D patients have hypertension, which amplifies vascular damage
- Diabetic dyslipidemia: ↑ triglycerides, ↑ LDL, ↓ HDL
Diabetic Nephropathy
- Leading cause of end-stage renal disease in the USA
- 30-40% of all diabetic patients develop clinical nephropathy
- Earliest sign: microalbuminuria (30-300 mg/day urinary albumin)
- Progresses to macroalbuminuria → hypertension → ESRD
- Morphology: glomerular basement membrane thickening, diffuse and nodular glomerulosclerosis (Kimmelstiel-Wilson nodules)
Diabetic Ocular Disease
- 60-80% of patients develop diabetic retinopathy - leading cause of adult blindness in the USA
- Key lesion: neovascularization driven by VEGF
- Also: cataracts (sorbitol), glaucoma
Diabetic Neuropathy
- Most common complication: peripheral neuropathy (symmetric, distal sensorimotor)
- Also: autonomic neuropathy (gastroparesis, orthostatic hypotension, impotence)
- Affects up to 50% overall; up to 80% of those with disease >15 years
Infections
- Impaired neutrophil function and poor tissue perfusion increase susceptibility
- Foot infections → gangrene → leading cause of nontraumatic limb amputation
Acute Metabolic Complications
| Feature | Diabetic Ketoacidosis (DKA) | Hyperosmolar Hyperglycemic State (HHS) |
|---|---|---|
| Type | Mainly T1D | Mainly T2D |
| Mechanism | Absolute insulin deficiency → lipolysis → ketones | Severe dehydration + hyperglycemia without significant ketosis |
| Blood glucose | Usually 250-600 mg/dL | Often >600 mg/dL |
| Ketones | Strongly positive | Minimal |
| pH | <7.3 | Usually normal |
| Risk | Coma, death | Coma, death |
Morphology Summary
| Type | Pancreatic Changes |
|---|---|
| T1D | Insulitis (lymphocytic infiltrate), marked reduction in islet cell mass |
| T2D | Amyloid deposition in islets (IAPP/islet amyloid polypeptide), mild reduction in β-cell mass |
Classic Triad of Symptoms
| Symptom | Mechanism |
|---|---|
| Polyuria (excess urination) | Glucosuria → osmotic diuresis |
| Polydipsia (excess thirst) | Dehydration from osmotic diuresis |
| Polyphagia (excess hunger) | Impaired glucose utilization at cellular level |
| Weight loss (especially T1D) | Catabolism - muscle/fat breakdown for energy |
| Blurred vision | Osmotic changes in lens |
Key Summary
- T1D: Autoimmune → absolute insulin deficiency → requires exogenous insulin
- T2D: Insulin resistance + relative insulin deficiency → begins with lifestyle/pharmacotherapy, may eventually need insulin
- Complications arise from chronic hyperglycemia damaging blood vessels and nerves via AGEs, PKC activation, and oxidative stress
- Severity of complications correlates directly with both the degree and duration of hyperglycemia - tight glycemic control demonstrably reduces end-organ damage
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 24, pp. 997-1010
This is a shared conversation. Sign in to Orris to start your own chat.