Edema types and pathogenesis
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have comprehensive information. Let me compile the full answer.
Edema: Types and Pathogenesis
Edema is the abnormal accumulation of excess fluid in the interstitial space of tissues or body cavities. About 60% of lean body mass is water - two-thirds intracellular, the remainder mostly interstitial, with only 5% in blood plasma. Fluid movement between the vascular and interstitial compartments obeys Starling's forces, and disruption of any one of them can cause edema.
Normal Starling Forces (Baseline)
Fluid movement across capillary walls is governed by four forces:
| Force | Direction | Effect |
|---|---|---|
| Capillary hydrostatic pressure (P_c) | Outward (filtration) | Pushes fluid out |
| Plasma colloid osmotic (oncotic) pressure (π_p) | Inward (reabsorption) | Pulls fluid in |
| Interstitial hydrostatic pressure (P_if) | Inward (minor) | Opposes filtration |
| Interstitial oncotic pressure (π_if) | Outward (minor) | Draws fluid out |
At the arteriolar end, net outward force exceeds inward force slightly, so a small amount of fluid leaks into the interstitium. Most of this is reabsorbed at the venular end. The residual is returned to circulation via lymphatics through the thoracic duct. Edema results when this balance is disrupted beyond the lymphatic drainage capacity.
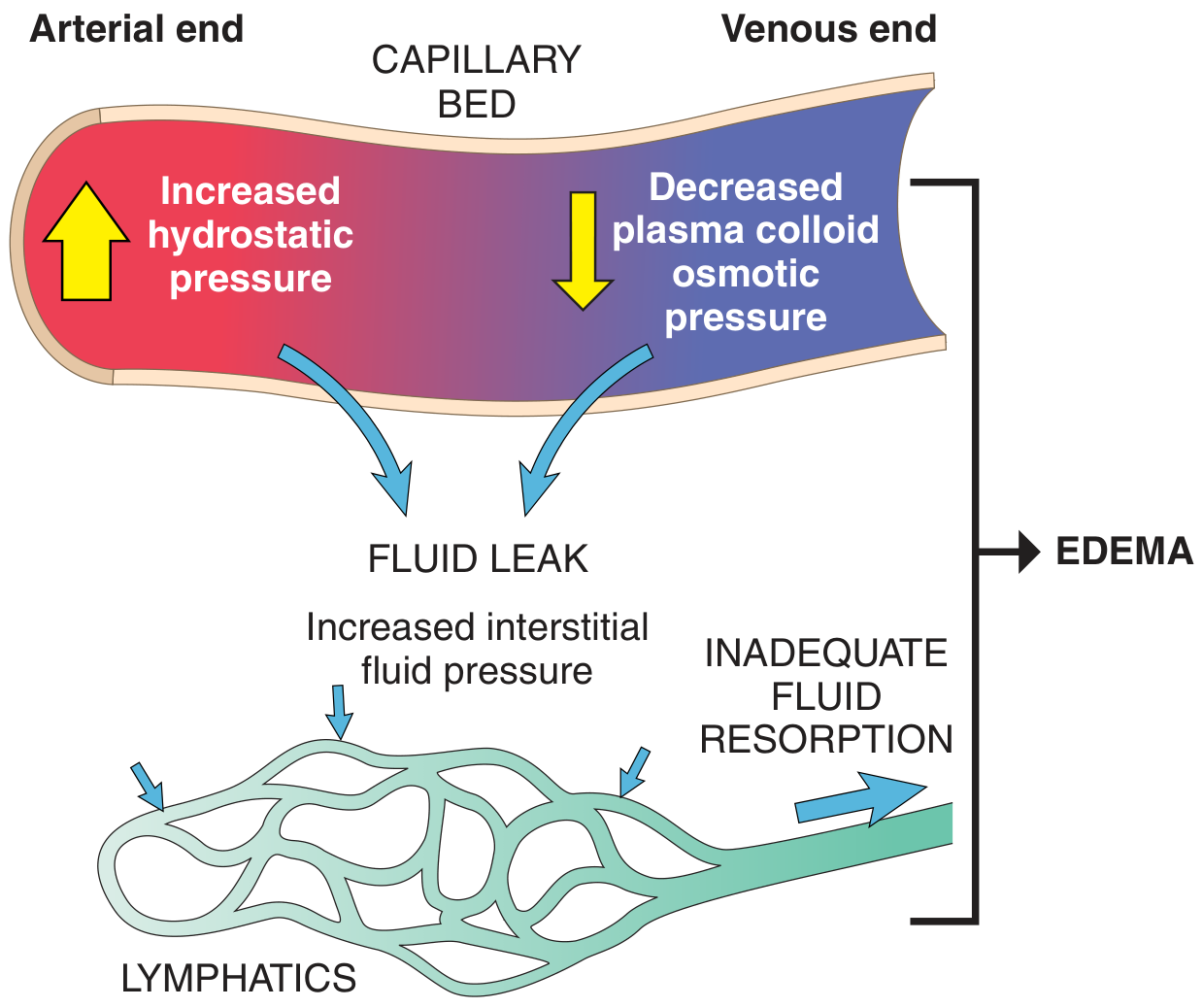
Fig. 3.2 - Factors influencing fluid movement across capillary walls (Robbins & Kumar Basic Pathology)
Four Primary Mechanisms of Edema Formation
1. Increased Hydrostatic Pressure
Mechanism: Elevated venous pressure is transmitted back to the capillaries, raising P_c and driving more fluid into the interstitium than lymphatics can drain.
Causes:
- Impaired venous return:
- Congestive heart failure (most common systemic cause)
- Constrictive pericarditis
- Liver cirrhosis (portal hypertension)
- Deep vein thrombosis (DVT) - causes unilateral, pitting, painful edema
- Venous obstruction by mass or tumor
- Prolonged dependency (long-distance travel, immobility)
- Arteriolar dilation:
- Heat
- Neurohumoral dysregulation
Characteristics of resulting edema: Dependent, pitting, protein-poor transudate. Unilateral (DVT) or bilateral (heart failure, renal failure, cirrhosis).
Note: Systemic arterial hypertension does NOT cause peripheral edema because precapillary sphincter autoregulation prevents arterial pressure from being transmitted to capillaries.
Heart failure pathogenesis in detail:
- Reduced cardiac output → venous pooling → increased capillary P_c
- Reduced cardiac output → renal hypoperfusion → activation of renin-angiotensin-aldosterone system (RAAS) → secondary hyperaldosteronism → Na⁺ and H₂O retention → expanded blood volume → worsening edema (vicious cycle)
2. Reduced Plasma Osmotic (Oncotic) Pressure
Mechanism: Low plasma albumin reduces the oncotic "pull" that normally reabsorbs fluid at the venular end. More fluid leaks out and less returns, overwhelming lymphatics.
Causes (all lead to hypoalbuminemia):
- Nephrotic syndrome (most important) - glomerular damage allows albumin to spill into urine (albuminuria)
- Liver disease (cirrhosis, advanced liver failure) - reduced hepatic albumin synthesis
- Malnutrition / kwashiorkor - protein deficiency impairs albumin production
- Protein-losing enteropathy - GI protein loss exceeds synthesis
Pathogenesis of nephrotic syndrome edema:
Low albumin → decreased oncotic pressure → edema → reduced intravascular volume → renal hypoperfusion → RAAS activation → Na⁺ and H₂O retention → increased blood volume (but protein still low) → worsening edema. A self-perpetuating cycle: increased salt and water retention cannot fix the root defect (low albumin) and only makes edema worse.
Characteristics: Bilateral, pitting transudate. Nephrotic edema tends to appear first in loose connective tissue (periorbital edema, eyelids) before becoming dependent.
3. Lymphatic Obstruction (Lymphedema)
Mechanism: Even when Starling forces are normal, the ~10% of capillary filtrate that is not reabsorbed at the venular end must be removed by lymphatics. Obstruction of lymphatic drainage causes protein-rich fluid to accumulate in the interstitium.
Causes:
- Infectious: Filariasis (Wuchereria bancrofti) - fibrosis of inguinal lymphatics and lymph nodes → elephantiasis of lower extremity and genitalia
- Neoplastic: Tumor infiltration or compression of lymphatics; breast cancer → peau d'orange (orange peel skin appearance from cutaneous lymphatic obstruction)
- Postsurgical: Axillary lymph node dissection (breast cancer surgery) → arm lymphedema
- Postirradiation: Radiation fibrosis of lymphatics
Characteristics: Non-pitting (because the protein-rich fluid stimulates fibrosis over time), localized. Chronic lymphedema leads to tissue fibrosis and skin thickening.
4. Increased Vascular Permeability (Inflammatory Edema)
Mechanism: Inflammatory mediators (histamine, bradykinin, leukotrienes, prostaglandins, VEGF, substance P) cause endothelial cell contraction, widening of intercellular junctions, and direct endothelial injury. This allows protein-rich plasma to leak into the interstitium.
Causes:
- Acute inflammation (infections, trauma, burns, allergic reactions)
- Chronic inflammation
- Angiogenesis (new, leaky vessels)
- ARDS (acute respiratory distress syndrome)
Characteristics: Produces a protein-rich exudate (not transudate). Localized to site of inflammation. The high protein content of the exudate raises interstitial oncotic pressure, which perpetuates fluid accumulation. Also associated with heat, redness, pain.
Transudate vs. Exudate:
| Feature | Transudate | Exudate |
|---|---|---|
| Protein content | Low (<3 g/dL) | High (>3 g/dL) |
| Specific gravity | <1.012 | >1.020 |
| Mechanism | Hydrostatic / oncotic imbalance | Increased permeability |
| Cause | CHF, nephrotic, cirrhosis | Infection, inflammation |
5. Sodium and Water Retention
Mechanism: Excessive renal retention of Na⁺ (and obligate water) expands intravascular volume, raising capillary hydrostatic pressure AND diluting plasma proteins (lowering oncotic pressure) - a dual hit.
Causes:
- Renal failure (acute or chronic)
- Excessive salt intake with impaired renal excretion
- Secondary hyperaldosteronism (from CHF, cirrhosis, nephrotic syndrome)
- Poststreptococcal glomerulonephritis
Summary Flowchart: Systemic Edema
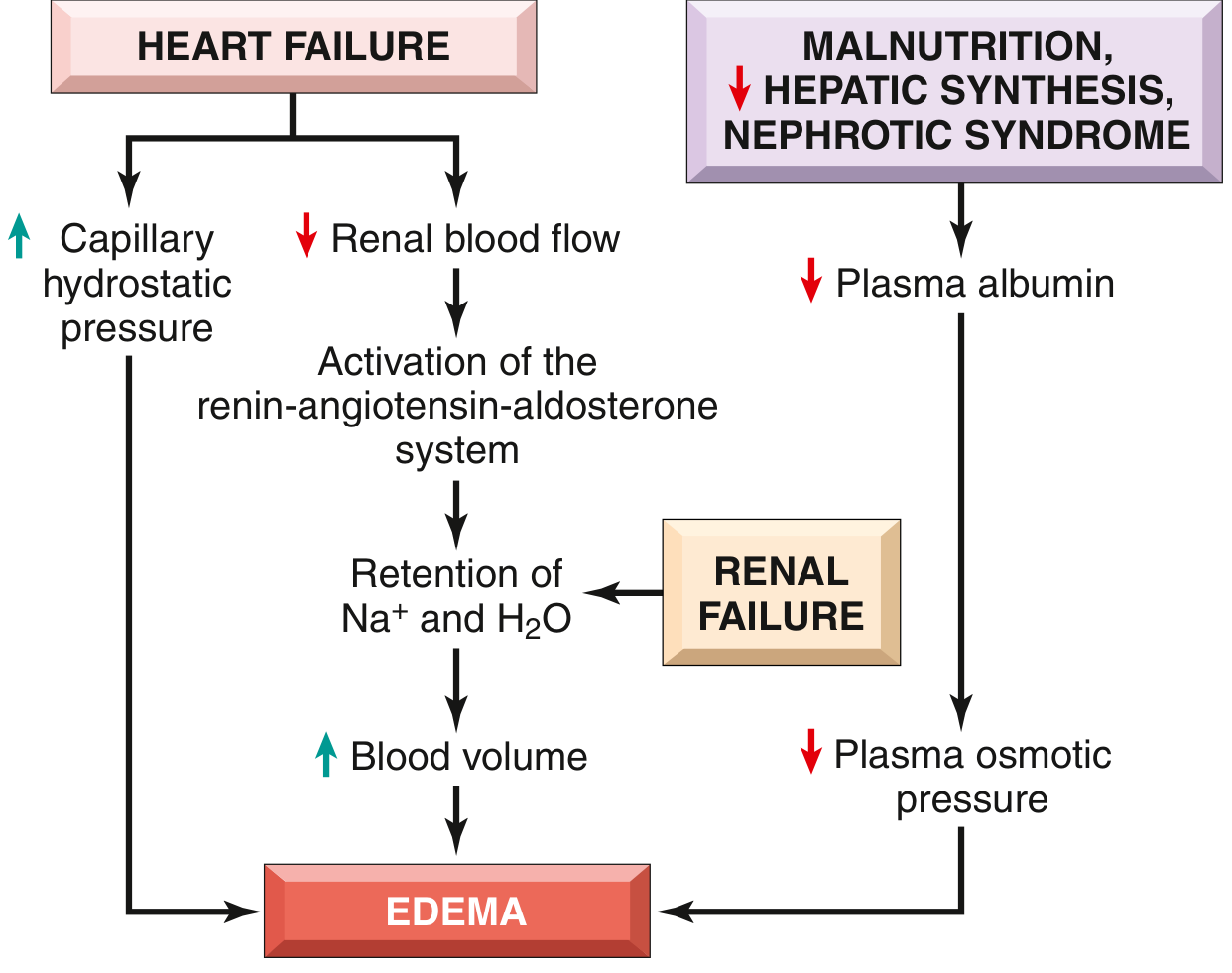
Fig. 3.3 - Pathways leading to systemic edema (Robbins & Kumar Basic Pathology)
Morphological Consequences by Location
| Site | Features |
|---|---|
| Subcutaneous | Dependent (legs when standing, sacrum when recumbent); pitting edema when pressed; periorbital in nephrotic syndrome |
| Pulmonary | Lungs 2-3x normal weight; frothy, blood-tinged fluid on cut surface; initially basal (gravity); "heart failure cells" = hemosiderin-laden alveolar macrophages from phagocytosed RBCs |
| Brain | Narrowed sulci, flattened gyri against skull; can cause herniation and death |
| Liver (chronic congestion) | "Nutmeg liver" - centrilobular red-brown necrosis surrounded by yellow viable tissue |
Mechanisms of Edema Formation (Diagrammatic)
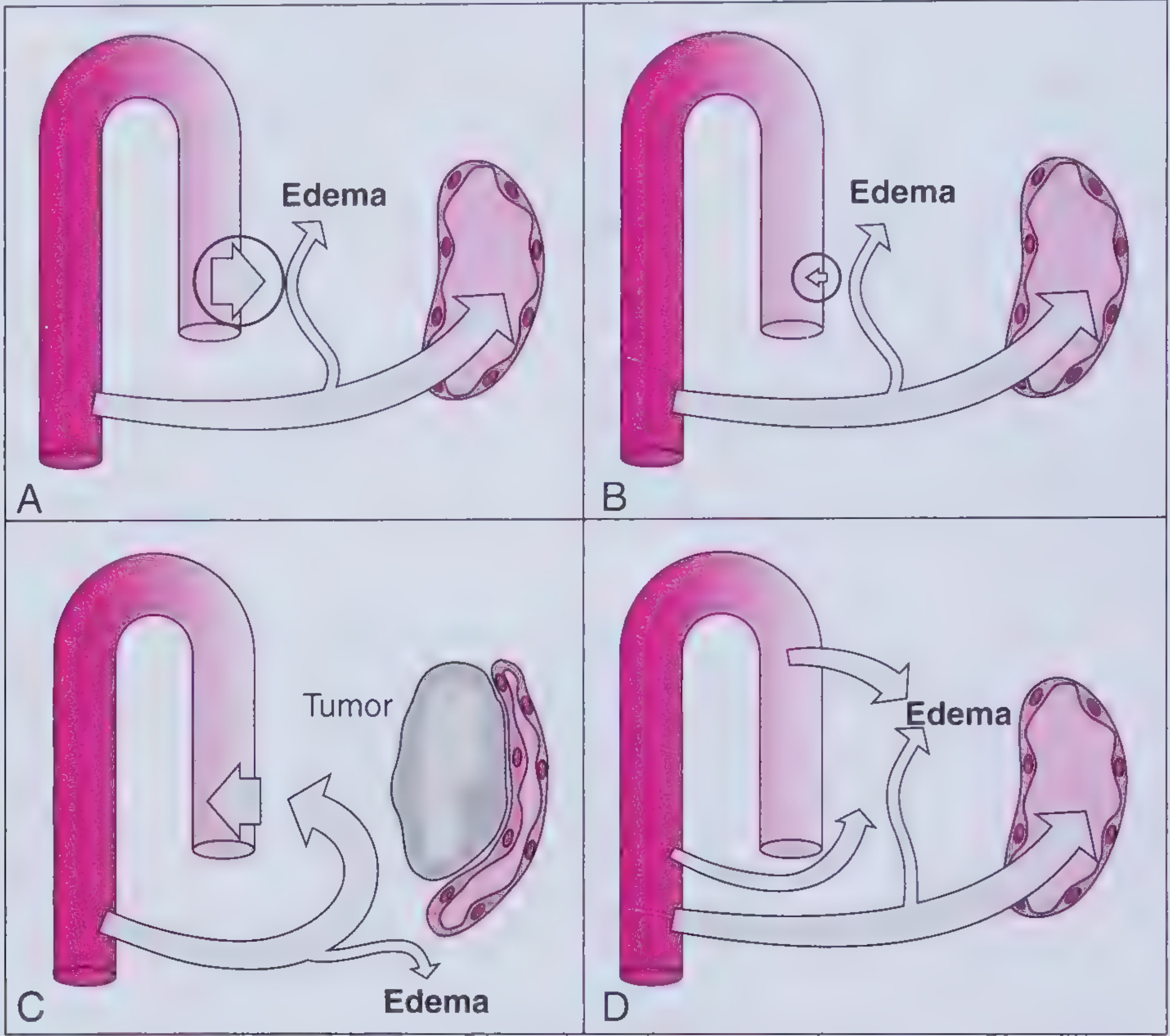
A = Increased capillary hydrostatic pressure | B = Decreased capillary oncotic pressure | C = Increased interstitial oncotic pressure (lymphatic obstruction) | D = Increased capillary permeability (Frameworks for Internal Medicine)
Special Named Edema Types
| Name | Definition |
|---|---|
| Anasarca | Severe generalized edema with profound subcutaneous swelling + fluid in all body cavities |
| Hydrothorax | Fluid in pleural cavity |
| Hydropericardium | Fluid in pericardial cavity |
| Ascites / Hydroperitoneum | Fluid in peritoneal cavity |
| Elephantiasis | Massive lower extremity / genital lymphedema from filarial infection |
| Peau d'orange | Orange peel skin from cutaneous lymphatic obstruction in breast cancer |
| Cerebral edema | Can be vasogenic (BBB breakdown) or cytotoxic (cell swelling) |
| Pulmonary edema | Cardiogenic (hydrostatic) vs. non-cardiogenic (ARDS, permeability) |
| Myxedema | Non-pitting edema from hypothyroidism; accumulation of hydrophilic glycosaminoglycans in dermis increases interstitial oncotic pressure |
Sources: Robbins & Kumar Basic Pathology (10e), p. 78-81 | Robbins, Cotran & Kumar Pathologic Basis of Disease | Frameworks for Internal Medicine, Ch. 23
Important questions from general pathology
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
I now have enough comprehensive material from Robbins to compile a thorough long-essay question guide covering all of general pathology. Let me write the complete answer.
Important Long Essay Questions - General Pathology
Based on Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease, here are the most important and frequently examined long essay questions, organized by topic with key answer points for each.
UNIT 1: CELL INJURY, DEATH & ADAPTATIONS
Q1. Classify cell injury. Describe the mechanisms of cell injury with special reference to ischemic/hypoxic injury.
Key points to cover:
Classification of cell injury:
- By reversibility: Reversible vs. Irreversible
- By cause: Physical, chemical, biological, immunological, nutritional, genetic
Causes of cell injury (mnemonic: CHIN-PROG):
Chemicals, Hypoxia/ischemia, Infections, Nutritional, Physical, Radiation, Oxygen free radicals, Genetic
Sequence of events in ischemic cell injury:
- Reduced O2 → impaired oxidative phosphorylation → ATP depletion
- ATP depletion → failure of Na⁺-K⁺-ATPase → cellular swelling (hydropic change)
- Anaerobic glycolysis → lactic acid → pH drop → clumping of nuclear chromatin
- Ribosomes detach from ER → reduced protein synthesis
- Lipid deposition (free fatty acids accumulate)
- Point of no return (irreversible injury):
- Mitochondrial vacuolization (large densities appear)
- Massive Ca²⁺ influx
- Plasma membrane damage
- Lysosomal rupture → autolysis
- Cell death
Key mechanisms of cell injury:
- ATP depletion - most common consequence of ischemia
- Mitochondrial damage - leads to cytochrome c release, apoptosis cascade
- Intracellular Ca²⁺ accumulation - activates phospholipases, proteases, endonucleases, ATPases
- Free radical injury / Reactive oxygen species (ROS):
- Sources: radiation, reperfusion of ischemic tissue, inflammation, metabolism
- Types: superoxide (O₂⁻), H₂O₂, hydroxyl radical (OH•)
- Damage: lipid peroxidation, protein modification, DNA damage
- Defense: SOD, catalase, glutathione peroxidase, vitamins C and E
- Defects in membrane permeability - loss of selective permeability
- DNA damage
Morphology of reversible injury:
- Cellular swelling (most common, first change)
- Fatty change (steatosis - liver, heart, kidney)
- Plasma membrane blebs
- EM: mitochondrial swelling, ER dilation, ribosome detachment
Q2. Define necrosis. Describe the types of necrosis with examples and their significance.
Definition: Necrosis is cell death accompanied by denaturation and/or digestion of dead cells, with associated inflammatory reaction.
Nuclear changes (triad):
- Pyknosis - nuclear shrinkage and increased basophilia (DNA condenses)
- Karyorrhexis - nuclear fragmentation
- Karyolysis - fading basophilia from DNase digestion
Cytoplasmic changes: Increased eosinophilia, glassy homogeneous appearance, vacuolation ("moth-eaten"), myelin figures
Types of necrosis:
| Type | Mechanism | Location | Gross Appearance | Microscopy |
|---|---|---|---|---|
| Coagulative | Ischemia; protein denaturation preserves cell outline | All solid organs except brain (classic = infarct) | Firm, pale, wedge-shaped | Anucleate "ghost" cells, tissue architecture preserved for days |
| Liquefactive | Bacterial/fungal infection; hypoxic death of CNS | Brain (infarct), abscesses | Softening, liquid, creamy pus | Complete digestion of cells, cavity formation |
| Caseous | Tuberculous infection (cell-mediated immunity) | Lymph nodes, lungs (TB) | Cheese-like, friable, yellow-white | Amorphous granular pink debris; no architecture; surrounded by granuloma |
| Fat necrosis | Pancreatic enzyme release; trauma to fat | Peritoneal fat, breast | Chalky white spots (saponification) | Shadowy fat cell outlines + basophilic calcium deposits + inflammation |
| Fibrinoid | Immune complex deposition in vessel walls; malignant hypertension | Blood vessel walls | Not grossly visible | Bright pink, amorphous, fibrin-like material in vessel walls |
| Gangrenous | Ischemia + bacterial superinfection of limb | Lower extremity | Dry (coagulative) or Wet (liquefactive) | Features of coagulative ± liquefactive |
Q3. Define apoptosis. Describe the mechanisms (pathways) of apoptosis and compare with necrosis.
Definition: Apoptosis is programmed, energy-dependent cell death characterized by cell shrinkage, chromatin condensation, membrane blebbing, and fragmentation into apoptotic bodies - without inflammation.
Physiological examples: Embryogenesis, hormone-dependent involution (endometrium), deletion of self-reactive lymphocytes, cell death after cytotoxic T-cell killing
Pathological examples: Cell death in DNA damage, viral infection, parenchymal cell death in organ obstruction, tumor necrosis
Pathways:
1. Intrinsic (Mitochondrial) Pathway:
- Stimulus: DNA damage, growth factor withdrawal, protein misfolding, oxidative stress
- BCL-2 family: Pro-apoptotic (BAX, BAK) vs. anti-apoptotic (BCL-2, BCL-XL)
- When apoptotic signals dominate, BAX/BAK form pores in mitochondrial membrane
- Cytochrome c leaks into cytosol → binds APAF-1 → activates caspase-9 → executioner caspases (3, 6, 7)
- P53 is a key inducer of this pathway via upregulation of BAX
2. Extrinsic (Death Receptor) Pathway:
- TNF receptor-1 (TNFR1) or Fas (CD95) binds TNF or FasL (on cytotoxic T cells)
- Death domain recruits adaptor proteins → activates caspase-8 → executioner caspases
- Key role: elimination of autoreactive lymphocytes (FasL/Fas); CTL killing
Clearance: Phosphatidylserine flips to outer leaflet of membrane → "eat-me" signal for macrophages. No inflammation - this is a defining feature.
Necrosis vs. Apoptosis:
| Feature | Necrosis | Apoptosis |
|---|---|---|
| Cell size | Swelling | Shrinkage |
| Nucleus | Pyknosis, karyorrhexis, karyolysis | Condensation, fragmentation |
| Plasma membrane | Disrupted | Intact (forms blebs) |
| Contents | Leaked (inflammation) | Packaged in apoptotic bodies |
| Inflammation | YES | NO |
| Energy requirement | Not needed (passive) | Requires ATP |
| Physiologic or pathologic | Always pathologic | Both |
Other cell death types: Necroptosis (TNF-induced, features of both), Pyroptosis (inflammasome-induced, IL-1 release, fever), Ferroptosis (iron-dependent)
Q4. Describe the cellular adaptations to stress - hypertrophy, hyperplasia, atrophy, metaplasia.
Hypertrophy = Increase in cell SIZE (no new cells)
- Mechanism: Increased production of organelles and proteins; stretch/growth factors activate signaling
- Examples: Left ventricular hypertrophy (pressure overload), uterine hypertrophy in pregnancy, skeletal muscle after exercise
- Pathological: Hypertrophied heart eventually decompensates (concentric → dilated)
Hyperplasia = Increase in cell NUMBER (through cell division; only in cells capable of dividing)
- Physiological: Liver regeneration, breast glandular tissue in pregnancy, bone marrow in anemia
- Hormonal: Endometrial hyperplasia (excess estrogen)
- Compensatory: Remaining kidney after nephrectomy
- Pathological: BPH, nodular hyperplasia of adrenal cortex
Atrophy = Decrease in cell SIZE and function (or loss of cells)
- Mechanisms: Decreased protein synthesis + increased protein degradation (ubiquitin-proteasome system, autophagy)
- Types: Disuse (immobilization), denervation, loss of endocrine stimulation, ischemia, malnutrition, pressure (tumor atrophy)
- Morphology: Brown atrophy - lipofuscin accumulation in atrophic cells (heart, liver)
Metaplasia = Replacement of one differentiated cell type by another - an adaptive response to chronic stress
- Always reversible if stimulus removed
- Mechanism: Reprogramming of stem cells; driven by growth factors, cytokines, matrix components
- Examples:
- Squamous metaplasia: Columnar → squamous in bronchi (smokers); endocervix (chronic irritation); bladder (stones)
- Intestinal metaplasia: Gastric mucosa in chronic gastritis (Barrett's esophagus: squamous → intestinal columnar)
- Significance: Pre-malignant potential (Barrett's → esophageal adenocarcinoma; bronchial squamous metaplasia → squamous carcinoma)
Dysplasia = Disordered growth with atypical cells - NOT a true adaptation; represents pre-neoplastic change; graded mild/moderate/severe
UNIT 2: INFLAMMATION
Q5. Define acute inflammation. Describe its vascular and cellular events, chemical mediators, and outcomes.
Definition: Acute inflammation is the rapid, stereotyped response of vascularized tissue to infection or tissue injury, characterized by exudation of fluid and plasma proteins, and emigration of leukocytes (predominantly neutrophils).
Purpose: Deliver leukocytes and plasma proteins to the site of injury to destroy the agent and initiate repair.
VASCULAR EVENTS (Triple Response of Lewis):
- Transient vasoconstriction (seconds)
- Vasodilation (arterioles then capillaries) - due to histamine, nitric oxide → redness (rubor) and heat (calor)
- Increased vascular permeability → fluid exudation → swelling (tumor)
- Stasis - slowing of blood flow as fluid leaves
Mechanisms of increased vascular permeability:
- Endothelial contraction (histamine, bradykinin, leukotrienes) - MOST COMMON; affects venules
- Direct endothelial injury (burns, toxins)
- Leukocyte-mediated injury
- New vessel formation (angiogenesis - leaky vessels)
CELLULAR EVENTS (Neutrophil emigration - 4 steps):
- Margination and Rolling - slowing of flow + selectin-mediated loose adhesion; P-selectin, E-selectin on endothelium bind sialyl-Lewis X on neutrophils
- Adhesion (firm) - ICAM-1/VCAM-1 (upregulated by TNF, IL-1) bind integrins (LFA-1, Mac-1) on neutrophils
- Transmigration (Diapedesis) - neutrophils squeeze between endothelial cells; PECAM-1 (CD31) on both
- Chemotaxis - directional migration toward highest concentration of chemoattractants: C5a, LTB4, IL-8 (CXCL8), bacterial peptides (N-formyl-methionyl peptides)
Phagocytosis:
- Recognition (aided by opsonins: IgG, C3b)
- Engulfment (pseudopod formation)
- Intracellular killing:
- Oxygen-dependent: NADPH oxidase → superoxide → H₂O₂ → hypochlorous acid (HOCl via myeloperoxidase - most potent)
- Oxygen-independent: lysozyme, defensins, elastase, BPI, lactoferrin
CHEMICAL MEDIATORS:
| Mediator | Source | Main Effect |
|---|---|---|
| Histamine | Mast cells, basophils, platelets | Vasodilation, vascular permeability |
| Serotonin | Platelets | Vasodilation, permeability |
| Prostaglandins (PGE2, PGI2) | Arachidonic acid (COX pathway) | Vasodilation, pain, fever |
| Leukotrienes (LTB4) | Arachidonic acid (LOX pathway) | Chemotaxis; LTC4/D4/E4 → bronchoconstriction, permeability |
| PAF | Mast cells, leukocytes, endothelium | Permeability, leukocyte activation, platelet aggregation |
| TNF, IL-1 | Macrophages | Systemic (fever, acute phase), endothelial activation (adhesion molecules) |
| IL-8 (CXCL8) | Macrophages, endothelium | Chemotaxis of neutrophils |
| IL-6 | Macrophages | Acute phase response |
| Complement (C3a, C5a) | Plasma | C3a/C5a = anaphylatoxins (mast cell degranulation); C5a = chemotaxis; C3b = opsonin |
| Bradykinin | Plasma (kinin system) | Permeability, pain, vasodilation |
| Nitric oxide (NO) | Endothelium, macrophages | Vasodilation, killing of microbes |
OUTCOMES of acute inflammation:
- Resolution - complete restoration (if minimal tissue destruction)
- Suppuration (abscess formation) - pus accumulates if intense pyogenic infection
- Organization and fibrosis - when tissue cannot regenerate or fibrin exudate not removed
- Progression to chronic inflammation - if agent persists
Q6. Define chronic inflammation. Describe its causes, cells involved, morphology, and outcomes. Compare with acute inflammation.
Definition: Chronic inflammation is inflammation of prolonged duration (weeks to months) characterized by tissue destruction, infiltration by mononuclear cells (macrophages, lymphocytes, plasma cells), and attempts at repair (angiogenesis, fibrosis) - all occurring simultaneously.
Causes:
- Persistent infections (Mycobacterium tuberculosis, fungi, parasites - organisms resistant to killing)
- Immune reactions (autoimmune diseases: rheumatoid arthritis, SLE)
- Prolonged exposure to toxic agents (silica → silicosis, smoking)
- Some infections from the start (viral hepatitis, H. pylori)
Cells involved:
- Macrophages - key cell; activated by IFN-γ (from T cells) or microbial products
- Secrete: proteases, ROS, NO (tissue destruction), TNF, IL-1, IL-12, growth factors
- "Angry macrophages" (epithelioid cells) form granulomas
- Lymphocytes - T cells (Th1 → IFN-γ activates macrophages; Th2 → IL-4,5,13 promote eosinophil/antibody response); B cells → plasma cells → antibodies
- Plasma cells - secrete antibodies; Russell bodies = immunoglobulin accumulation in cytoplasm
- Eosinophils - parasitic infections, IgE-mediated allergy; major basic protein causes tissue damage
- Mast cells - release histamine, cytokines
Morphology: Mononuclear infiltrate, new vessel formation (angiogenesis), fibrosis, tissue destruction (often ongoing simultaneously)
Granulomatous inflammation - specialized form of chronic inflammation:
- Granuloma = aggregate of activated macrophages (epithelioid cells) + lymphocytes, often with giant cells
- Giant cell types:
- Langhans giant cell (nuclei arranged in horseshoe/peripheral pattern) - TB, sarcoid
- Foreign body giant cell (nuclei randomly scattered centrally)
- Caseating granuloma (central caseous necrosis) - Tuberculosis, leprosy, fungi (Histoplasma)
- Non-caseating granuloma - Sarcoidosis, Crohn's disease, berylliosis, foreign bodies
- TB granuloma formation mechanism: M. tuberculosis phagocytosed by macrophage → incomplete killing → Th1 CD4+ cells → IFN-γ → macrophage activation → epithelioid transformation → granuloma
Q7. Describe the systemic effects of inflammation (Acute Phase Response) and the role of cytokines.
- Fever: IL-1, TNF, IL-6 → prostaglandin (PGE2) synthesis in hypothalamus → raises temperature set point; antipyretics (aspirin, ibuprofen) inhibit COX
- Acute phase proteins (liver synthesis stimulated by IL-6, IL-1, TNF):
- CRP (C-reactive protein) - opsonin, activates complement
- Fibrinogen - elevated ESR
- Serum amyloid A (SAA) - precursor to secondary amyloidosis
- Hepcidin - causes anemia of chronic disease (reduced iron availability)
- Negative acute phase reactants: albumin, transferrin (decrease)
- Leukocytosis: IL-1, TNF → bone marrow stimulation; bacterial = neutrophilia; viral = lymphocytosis; parasites/allergies = eosinophilia
- Lymphadenopathy: Macrophage and lymphocyte proliferation in draining nodes
- Septic shock (severe systemic): TNF, IL-1 → massive vasodilation, DIC, multi-organ failure
UNIT 3: HEMODYNAMIC DISORDERS
Q8. Describe the types and pathogenesis of edema. (Covered in detail in the previous session)
Q9. Describe the pathogenesis, types, fate, and complications of thrombus.
Virchow's Triad (causes of thrombosis):
- Endothelial injury - most important alone; exposes subendothelial collagen, releases vWF, tissue factor
- Stasis (abnormal blood flow) or turbulence - allows platelet/factor accumulation; atrial fibrillation, venous stasis, aneurysms
- Hypercoagulability (thrombophilia) - primary (factor V Leiden, antithrombin III deficiency, protein C/S deficiency) or secondary (smoking, cancer, pregnancy, HRT, immobility, DIC)
Types of thrombi:
- Arterial (white thrombus): Platelet-rich; lines of Zahn (alternating pale/red layers); occlusive; head attached to vessel wall
- Venous (red thrombus): Red cell-rich, fibrin network; extends proximally; DVT (leg veins most common)
- Mural thrombus: In cardiac chambers (post-MI, atrial fibrillation) or aortic aneurysm
- Valvular thrombus: Infective endocarditis (bacteria-laden, irregular vegetations); sterile vegetations in SLE (Libman-Sacks)
Fate of thrombus (4 Ds + Resolution):
- Dissolution (fibrinolysis) - ideal outcome; tissue plasminogen activator (tPA) activates plasmin
- Propagation - enlargement
- Organization and recanalization - ingrowth of endothelial cells, smooth muscle; partial restoration of flow
- Embolization - dislodged thrombus travels distally
- Calcification - phlebolith formation (venous thrombus)
Complications: Infarction (coronary = MI; cerebral = stroke); pulmonary embolism (from DVT); peripheral arterial occlusion; paradoxical embolism (patent foramen ovale)
Q10. Define infarction. Classify and describe the pathogenesis and morphology of infarcts.
Definition: Infarction is an area of ischemic necrosis in a tissue or organ resulting from occlusion of its arterial supply or venous drainage.
Types:
| Feature | Red (Hemorrhagic) Infarct | White (Pale/Anemic) Infarct |
|---|---|---|
| Mechanism | Venous occlusion OR arterial occlusion with collateral flow / reperfusion | Arterial occlusion in solid organs with end-arteries |
| Location | Lungs, small intestine, ovary, testis, brain (sometimes) | Heart, kidney, spleen |
| Appearance | Red-brown, hemorrhagic, soft | Pale, wedge-shaped, firm |
| Histology | Coagulative necrosis + RBC extravasation | Coagulative necrosis ("ghost cells") |
Pathogenesis: Arterial occlusion (atherosclerosis, embolism, thrombosis, vasospasm) → ischemia → ATP depletion → cell injury → necrosis (sequence as in Q1)
Factors determining severity: Rate of occlusion (gradual allows collateral development), presence of collateral circulation, oxygen-carrying capacity, tissue's vulnerability to hypoxia (neurons most sensitive - 3-5 min; myocardium - 20-30 min; skeletal muscle - hours)
UNIT 4: REPAIR AND REGENERATION
Q11. Describe wound healing - explain primary and secondary intention healing, the role of growth factors, and complications.
Labile, stable, and permanent cells:
- Labile (continuously dividing): Surface epithelia, GI mucosa, bone marrow - most regenerative capacity
- Stable (quiescent, can re-enter cycle): Hepatocytes, renal tubular cells, smooth muscle - regenerate after injury
- Permanent (non-dividing): Neurons, cardiac myocytes, skeletal muscle - NO regeneration; replaced by scar
Healing by primary intention (clean incised wound, edges apposed):
- Day 0-1: Clot forms; neutrophils appear at margins
- Day 2-3: Macrophages replace neutrophils; granulation tissue begins; keratinocytes migrate under clot
- Day 5-7: Granulation tissue fills wound; collagen fibers bridge gap; vascularity maximal
- Week 2: Collagen accumulation; fibroblast proliferation decreases; vascularity diminishes
- Month 1+: Scar tissue; tensile strength increases (but never exceeds 80% of unwounded skin)
Healing by secondary intention (large defect, irregular margins, infection):
- Same phases but: more granulation tissue, extensive wound contraction (myofibroblasts), larger scar
- Wound contraction = key difference; reduces wound area by ~80%
Key growth factors in wound healing:
- EGF (Epidermal Growth Factor): Keratinocyte/fibroblast proliferation
- TGF-α: Keratinocyte proliferation
- TGF-β: Key regulator; stimulates collagen synthesis; fibrosis if dysregulated
- PDGF: Fibroblast/smooth muscle recruitment; produced by platelets, macrophages
- FGF (basic): Angiogenesis, fibroblast stimulation
- VEGF: Angiogenesis (dominant signal)
- HGF (hepatocyte growth factor): Liver regeneration
Collagen synthesis and remodeling:
- Type III collagen (early) → replaced by Type I (late) by matrix metalloproteinases (MMPs) and TIMP balance
- Vitamin C required for hydroxylation of proline/lysine (scurvy → poor wound healing)
- Zinc required for MMP activity
Complications of wound healing:
- Wound dehiscence - suture failure or infection
- Keloid - exuberant collagen deposition extending beyond wound margins; more in dark-skinned individuals; type III collagen
- Hypertrophic scar - excess collagen within wound margins; regresses spontaneously
- Contracture - excessive contraction causing functional impairment (burns, digits)
- Proud flesh (excessive granulation tissue)
- Poor healing factors: Infection, foreign body, diabetes (impaired leukocyte function), malnutrition (protein, vitamin C, zinc deficiency), glucocorticoids, impaired blood supply
UNIT 5: NEOPLASIA
Q12. Define neoplasia. Classify tumors. Compare benign and malignant tumors.
Definition (Willis): "A neoplasm is an abnormal mass of tissue, the growth of which exceeds and is uncoordinated with that of the normal tissues and persists in the same excessive manner after cessation of the stimuli which evoked the change."
Benign vs. Malignant:
| Feature | Benign | Malignant |
|---|---|---|
| Differentiation | Well differentiated | Variable: well to undifferentiated (anaplastic) |
| Growth rate | Slow | Usually rapid |
| Mitoses | Rare, normal | Frequent, abnormal (tripolar, atypical) |
| Borders | Encapsulated, expansile | Invasive, no true capsule |
| Metastasis | NEVER | Yes (defining feature of malignancy) |
| Necrosis/hemorrhage | Rare | Common |
| Recurrence | Rare | Common |
| Effects | Local; pressure | Local + systemic |
| Nuclear features | Normal N:C ratio | High N:C ratio, pleomorphism, hyperchromatism, prominent nucleoli |
Nomenclature:
- Benign epithelial: Adenoma (glandular), papilloma, cystadenoma
- Malignant epithelial: Carcinoma (squamous cell, adenocarcinoma)
- Benign mesenchymal: Fibroma, lipoma, chondroma, osteoma, rhabdomyoma
- Malignant mesenchymal: Sarcoma (fibrosarcoma, liposarcoma, osteosarcoma)
- Mixed: Teratoma, Wilms' tumor, fibroadenoma
- Exception: Melanoma (malignant despite "-oma"); Lymphoma (malignant)
Q13. Describe the pathogenesis of carcinogenesis - oncogenes, tumor suppressor genes, and hallmarks of cancer.
Two-hit hypothesis (Knudson): Two mutations required to inactivate both alleles of tumor suppressor gene; first hit may be inherited (familial cancers) or somatic.
Proto-oncogenes → Oncogenes (gain of function; dominant):
- Mutations: Point mutation (RAS), amplification (HER2/neu, N-myc), translocation (BCR-ABL in CML; c-myc in Burkitt lymphoma)
- Functions: Growth factor receptors (EGFR), signal transducers (RAS), transcription factors (MYC), cell cycle regulators (Cyclin D1)
Tumor suppressor genes (loss of function; recessive):
- RB (retinoblastoma gene): G1/S checkpoint; phosphorylated (inactive) RB allows E2F to drive cell cycle; hypophosphorylated (active) RB blocks E2F
- TP53 ("guardian of the genome"): Responds to DNA damage → cell cycle arrest (p21 upregulation) or apoptosis (BAX upregulation); mutated in >50% of human cancers; Li-Fraumeni syndrome (germline mutation)
- APC: Colon cancer; regulates β-catenin; familial adenomatous polyposis (FAP) when germline
- BRCA1/BRCA2: DNA repair; breast and ovarian cancer (hereditary)
Hallmarks of Cancer (Hanahan & Weinberg):
- Sustaining proliferative signaling (oncogene activation)
- Evading growth suppressors (TSG loss)
- Resisting cell death (BCL-2 overexpression, p53 loss)
- Enabling replicative immortality (telomerase activation)
- Inducing angiogenesis (VEGF overproduction)
- Activating invasion and metastasis (E-cadherin loss, MMP upregulation)
- Reprogramming energy metabolism (Warburg effect - aerobic glycolysis)
- Evading immune destruction (PD-L1 expression)
Metastasis cascade:
Local invasion → intravasation → survival in circulation → arrest at distant site → extravasation → micrometastasis → colonization
- Loss of E-cadherin (epithelial-mesenchymal transition, EMT) is a key early step
- Common sites by cancer type: Lung, liver, bone (vertebral), brain, adrenal glands
UNIT 6: OTHER IMPORTANT TOPICS
Q14. Describe the pathogenesis, types, and consequences of amyloidosis.
- Definition: Extracellular deposits of abnormal, misfolded protein fibrils in β-pleated sheet configuration
- Staining: Congo red → apple-green birefringence under polarized light (pathognomonic)
- Types:
- Primary (AL amyloid): Immunoglobulin light chains; multiple myeloma/plasma cell dyscrasia; affects heart, kidney, tongue, nerves
- Secondary (AA amyloid): Serum amyloid A (SAA) protein; chronic inflammatory diseases (RA, osteomyelitis, TB, Crohn's); affects kidney, liver, spleen
- Hemodialysis-associated: β2-microglobulin; carpal tunnel syndrome
- Senile cardiac/systemic: Normal transthyretin (TTR); elderly patients
- Familial amyloid: Mutant TTR; hereditary
- Alzheimer's disease: Aβ peptide in plaques; Tau protein in neurofibrillary tangles
Q15. Describe the pathogenesis and classification of shock.
Definition: Shock is a state of systemic hypoperfusion that leads to inadequate oxygen delivery to meet metabolic demands, resulting in cellular dysfunction and, if untreated, cell death.
Types:
| Type | Mechanism | Key Cause | Hemodynamics |
|---|---|---|---|
| Hypovolemic | ↓ Blood/fluid volume | Hemorrhage, dehydration, burns | ↓CO, ↑SVR, ↑HR |
| Cardiogenic | Pump failure | MI, arrhythmia, tamponade | ↓CO, ↑SVR, ↑HR |
| Distributive (Septic) | Maldistribution of flow; vasodilation | Gram-negative sepsis (LPS) | ↑CO early/↓CO late, ↓SVR |
| Neurogenic | Loss of vasomotor tone | Spinal cord injury | ↓SVR, normal CO |
| Anaphylactic | IgE-mediated mast cell degranulation | Allergen | ↓SVR, bronchoconstriction |
Stages of shock:
- Compensated (non-progressive): Reflex mechanisms maintain perfusion (vasoconstriction, tachycardia, ADH, RAAS)
- Progressive (decompensated): Perfusion fails despite compensatory mechanisms; tissue hypoxia, lactic acidosis, cellular injury
- Irreversible: Cell death; multi-organ failure; death despite treatment
Septic shock pathogenesis: LPS/endotoxin → TLR4 → macrophage activation → massive TNF, IL-1, IL-6, IL-12 → endothelial activation → vasodilation (NO), DIC, increased permeability → distributive shock + ARDS + multi-organ failure
Quick Reference: High-Yield Topic Checklist
| Topic | Essay Type |
|---|---|
| Cell injury mechanisms (ischemic injury) | 10-15 mark long essay |
| Types of necrosis | 10 mark long essay |
| Apoptosis - pathways + comparison with necrosis | 10-15 mark long essay |
| Cellular adaptations (hypertrophy, hyperplasia, atrophy, metaplasia) | 10 mark |
| Acute inflammation (vascular + cellular + mediators) | 15 mark long essay |
| Chemical mediators of inflammation | 10 mark |
| Chronic inflammation + granulomatous inflammation | 10-15 mark |
| Wound healing (primary + secondary intention) | 10-15 mark |
| Edema types + pathogenesis | 10 mark |
| Thrombus - formation, types, fate, complications | 10 mark |
| Infarction - types, morphology | 5-10 mark |
| Carcinogenesis + oncogenes + tumor suppressor genes | 15 mark |
| Benign vs. malignant tumors (comparison) | 10 mark |
| Amyloidosis | 10 mark |
| Shock - types, pathogenesis | 10-15 mark |
| Free radical injury and antioxidant defenses | 5-10 mark |
| Calcification (dystrophic vs. metastatic) | 5 mark |
| Pigments (hemosiderin, bilirubin, lipofuscin, melanin) | 5 mark |
Sources: Robbins & Kumar Basic Pathology (10e) | Robbins, Cotran & Kumar Pathologic Basis of Disease (10e)
This is a shared conversation. Sign in to Orris to start your own chat.