A 50y old male complains of dragging sensation in abdomen. Physical examination revealed pallor and massive spleenomegaly. There is no lymphadenopathy. Total leucocyte count is 1,60,000 cell/mm³ and platelet count is 2 Lakhs/mm³. M a) What is the probable diagnosis? b) Describe cytogenetic change in this case? c) Enumerate peripheral blood smear findings? Give very brief answers pathology it for 5 marks question
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Here is a complete, exam-ready answer for a 5-mark question:
a) Probable Diagnosis: Chronic Myeloid Leukemia (CML)
Clues pointing to CML:
- Middle-aged male (peak incidence: 5th-6th decade)
- Dragging sensation in abdomen due to massive splenomegaly (a hallmark feature)
- Pallor (anemia from marrow infiltration)
- Markedly elevated TLC: 1,60,000/mm³ (leukocytosis is the defining feature)
- No lymphadenopathy (distinguishes from lymphoma/CLL)
- Platelets normal/elevated (2 lakhs) - typical in chronic phase CML
- CML is a myeloproliferative neoplasm (MPN), not a lymphoproliferative one
b) Cytogenetic Change: Philadelphia (Ph) Chromosome
Translocation: t(9;22)(q34;q11)
- A reciprocal translocation between chromosome 9 and chromosome 22
- The ABL proto-oncogene (on chromosome 9q34) translocates to the BCR (breakpoint cluster region) gene on chromosome 22q11
- This creates the Philadelphia (Ph) chromosome - a shortened chromosome 22
- Results in BCR::ABL fusion gene encoding a 210 kDa BCR-ABL chimeric protein
- BCR-ABL has constitutively active tyrosine kinase activity, driving growth factor-independent proliferation (via RAS, STAT, AKT pathways) with normal differentiation - hence the mature cells in peripheral blood
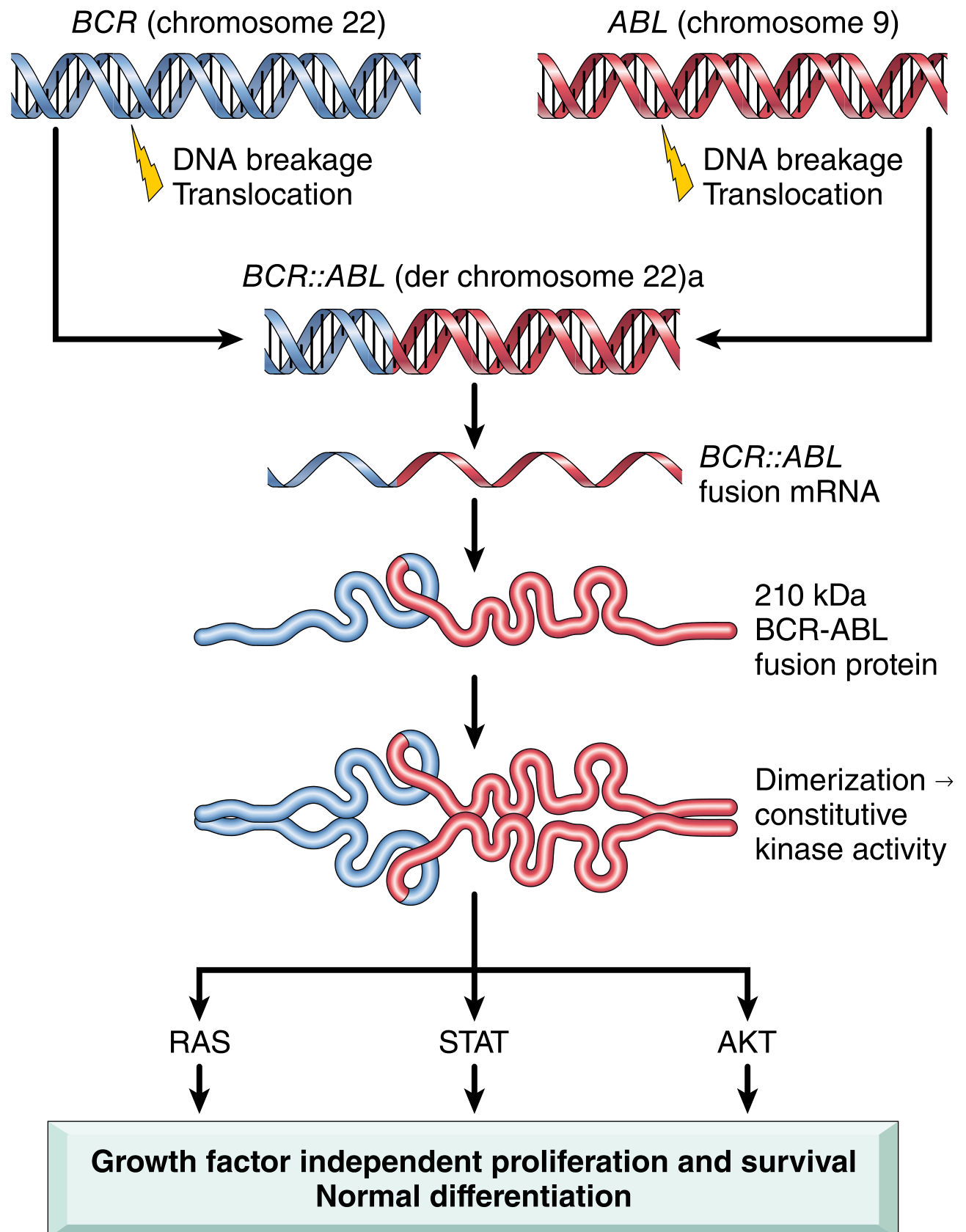
Present in >90% of CML cases; remaining cases have cryptic rearrangements detectable by FISH or PCR.
c) Peripheral Blood Smear Findings in CML
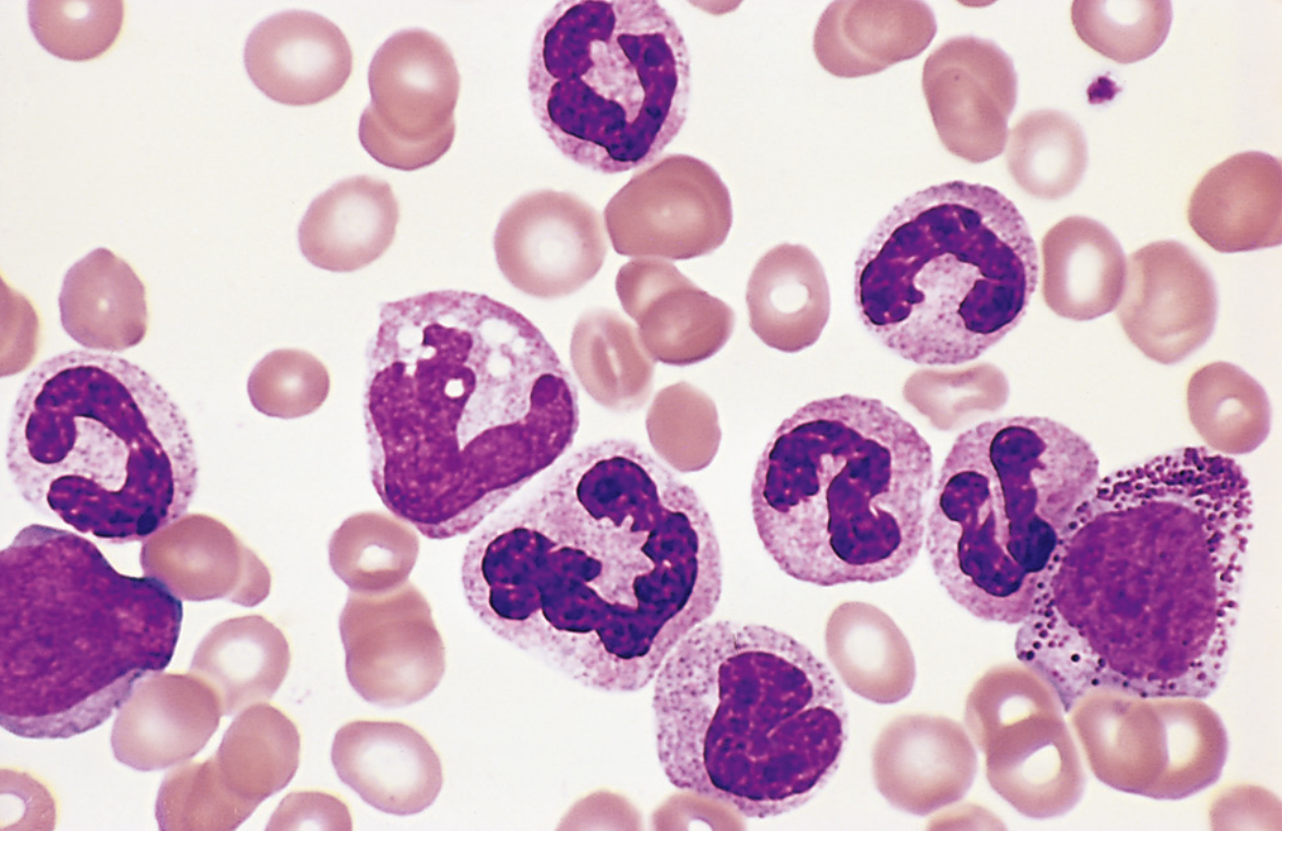
(Robbins & Kumar Pathologic Basis of Disease - Fig. 13.35: many mature neutrophils, metamyelocytes, and myelocytes)
| Finding | Details |
|---|---|
| Leukocytosis | WBC markedly elevated (often >1 lakh/mm³) |
| Full granulocytic spectrum | From myeloblasts to mature neutrophils - the "myelocyte bulge" (myelocytes + metamyelocytes predominate) |
| Myelocytes & metamyelocytes | Characteristic; more than any other stage |
| Basophilia | Increased basophils (virtually diagnostic of CML among MPNs) |
| Eosinophilia | Mild increase in eosinophils |
| Thrombocytosis | Increased/normal platelets with abnormal platelet morphology |
| Normocytic normochromic anemia | Pallor on RBC background |
| Blasts | <10% in chronic phase (if >20% = blast crisis) |
| Reduced/absent LAP score | Leukocyte Alkaline Phosphatase (LAP) score is low/zero (distinguishes from leukemoid reaction where LAP is high) |
Key diagnostic memory: The smear shows a "left shift" with the full myeloid maturation spectrum - myeloblast → promyelocyte → myelocyte (predominant) → metamyelocyte → band → neutrophil - along with basophilia and eosinophilia.
Sources: Robbins & Kumar Pathologic Basis of Disease (9780443264528); Goldman-Cecil Medicine (9780323930345)
13. Explain the laboratory approach to diagnose a male child with repeated joint bleeding. Pathology for 3 marks give briefly
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Here is a concise, exam-focused answer for 3 marks:
Laboratory Approach to Diagnose a Male Child with Repeated Joint Bleeding (Hemarthrosis)
Clinical clue: A male child with recurrent hemarthrosis points strongly to Hemophilia A (Factor VIII deficiency) or Hemophilia B (Factor IX deficiency) - both X-linked recessive disorders. The lab approach proceeds in two steps: screening, then confirmation.
Step 1 - Screening Tests
| Test | Expected Result in Hemophilia |
|---|---|
| Platelet count | Normal |
| Bleeding time (BT) | Normal |
| Prothrombin Time (PT/INR) | Normal (extrinsic pathway intact) |
| aPTT (activated Partial Thromboplastin Time) | Prolonged (intrinsic pathway defect) |
| Thrombin time | Normal |
- The hallmark pattern: Isolated prolonged aPTT with normal PT and normal platelet count/BT - this means the defect lies in the intrinsic pathway (Factors VIII, IX, XI, XII)
- Hemarthrosis specifically indicates a coagulation factor deficiency (deep tissue bleeding), not a platelet disorder
Step 2 - Specific Factor Assays (Confirmation)
Once aPTT is found prolonged in isolation, specific factor activity assays are done:
| Assay | Finding |
|---|---|
| Factor VIII activity assay | Reduced → Hemophilia A |
| Factor IX activity assay | Reduced → Hemophilia B |
Severity grading by factor level:
- Severe: <1% activity (spontaneous joint bleeds)
- Moderate: 1-5%
- Mild: 5-40%
Step 3 - Additional Tests
- Mixing study (1:1 with normal plasma): aPTT corrects → confirms factor deficiency (not inhibitor)
- vWF antigen & ristocetin cofactor assay: to distinguish from von Willebrand Disease (in vWD, Factor VIII may also be low but BT/PFA is abnormal and vWF is reduced)
- Genetic/molecular testing: to identify the causative mutation (useful for carrier detection and prenatal diagnosis)
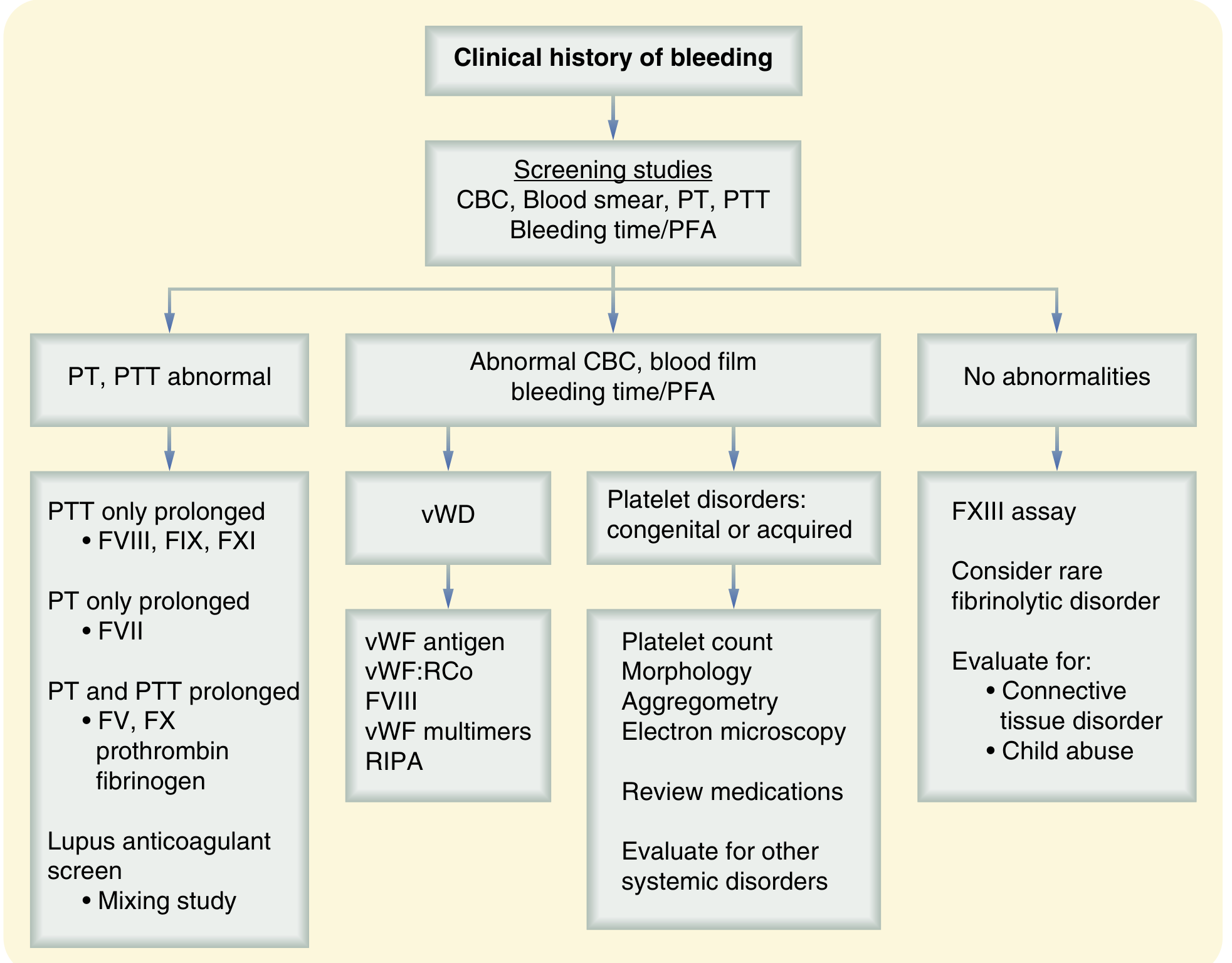
(Sabiston Textbook of Surgery - Lab evaluation flowchart: isolated prolonged PTT → check FVIII, FIX, FXI)
Summary for 3 marks:
- Screening: Platelet count + BT (normal), PT (normal), aPTT (prolonged) → intrinsic pathway defect
- Confirmation: Specific factor assays - Factor VIII ↓ = Hemophilia A; Factor IX ↓ = Hemophilia B
- Mixing study corrects aPTT (rules out inhibitor); vWF assay to exclude vWD
Sources: Goldman-Cecil Medicine (9780323930345); Sabiston Textbook of Surgery (9780443124341)
This is a shared conversation. Sign in to Orris to start your own chat.