Ocular manifestations of neurofibramotosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
neurofibromatosis ocular manifestations
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive information from Kanski's Clinical Ophthalmology (10th ed.), Fitzpatrick's Dermatology, and Goldman-Cecil Medicine. Here is the full answer:
Ocular Manifestations of Neurofibromatosis
Neurofibromatosis (NF) has two main forms - NF1 (von Recklinghausen disease) and NF2 - each with distinct but overlapping ocular findings. In NF2, ocular lesions are often the first manifestations of the disease.
Neurofibromatosis Type 1 (NF1)
Genetics: Autosomal dominant; NF1 gene (neurofibromin) on chromosome 17; ~50% new mutations. Prevalence 1:4000.
Eyelid
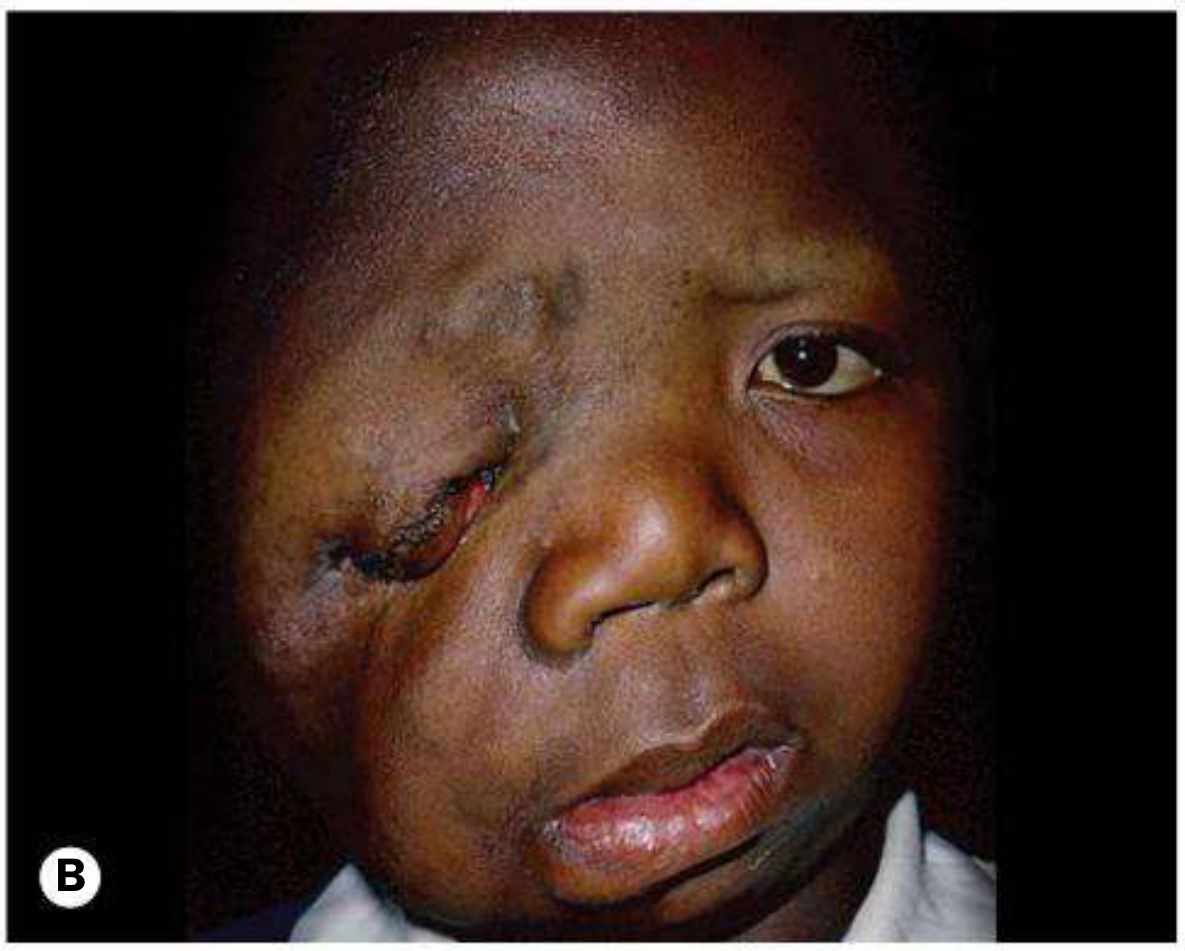
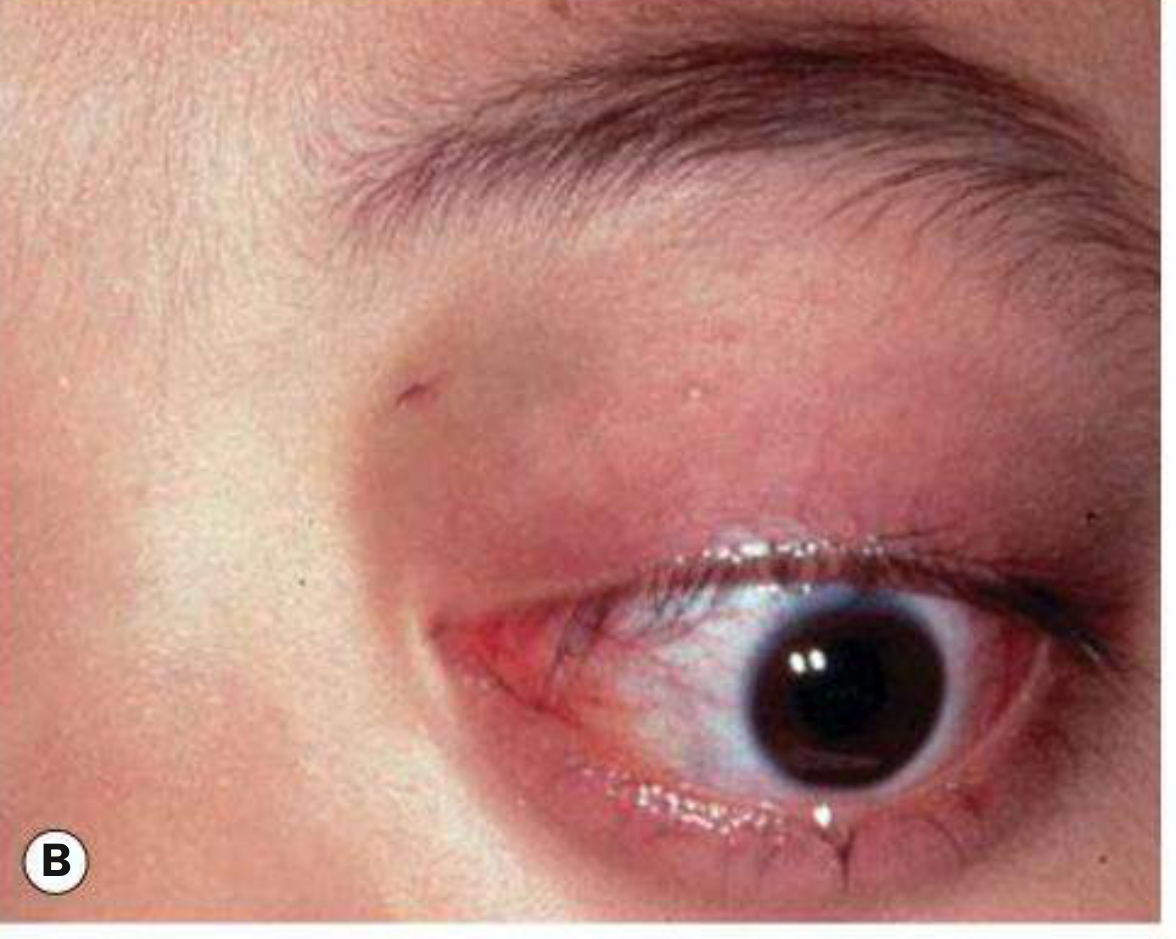
- Plexiform neurofibroma - the classic finding. Produces a characteristic S-shaped deformity of the upper eyelid. On palpation it feels like a "bag of worms." This is the eyelid lesion shown below:
Orbit
| Lesion | Key Features |
|---|---|
| Optic nerve glioma | 15-40% of patients; pilocytic astrocytoma; fusiform nerve enlargement; presents with painless proptosis, visual impairment (often marked), optic atrophy, strabismus; can extend to chiasm, optic tract, hypothalamus |
| Spheno-orbital encephalocoele | Absence of greater wing of sphenoid bone; causes pulsating proptosis |
| Other neural tumours | Neurilemmoma (schwannoma), plexiform neurofibroma, meningioma |
Iris
- Lisch nodules (at least 95%) - pathognomonic hamartomas appearing as tiny nodular pigmented lesions protruding above the iris surface. Develop during the 2nd-3rd decades. Both eyes are involved (bilateral).
- Congenital ectropion uveae - uncommon; may be associated with glaucoma
- Mammillations - rare
Cornea
- Prominent corneal nerves
Glaucoma
- Not common overall. When present, it is usually unilateral and congenital; ~50% of those cases have an ipsilateral upper eyelid neurofibroma and facial hemiatrophy.
Fundus
| Finding | Frequency / Notes |
|---|---|
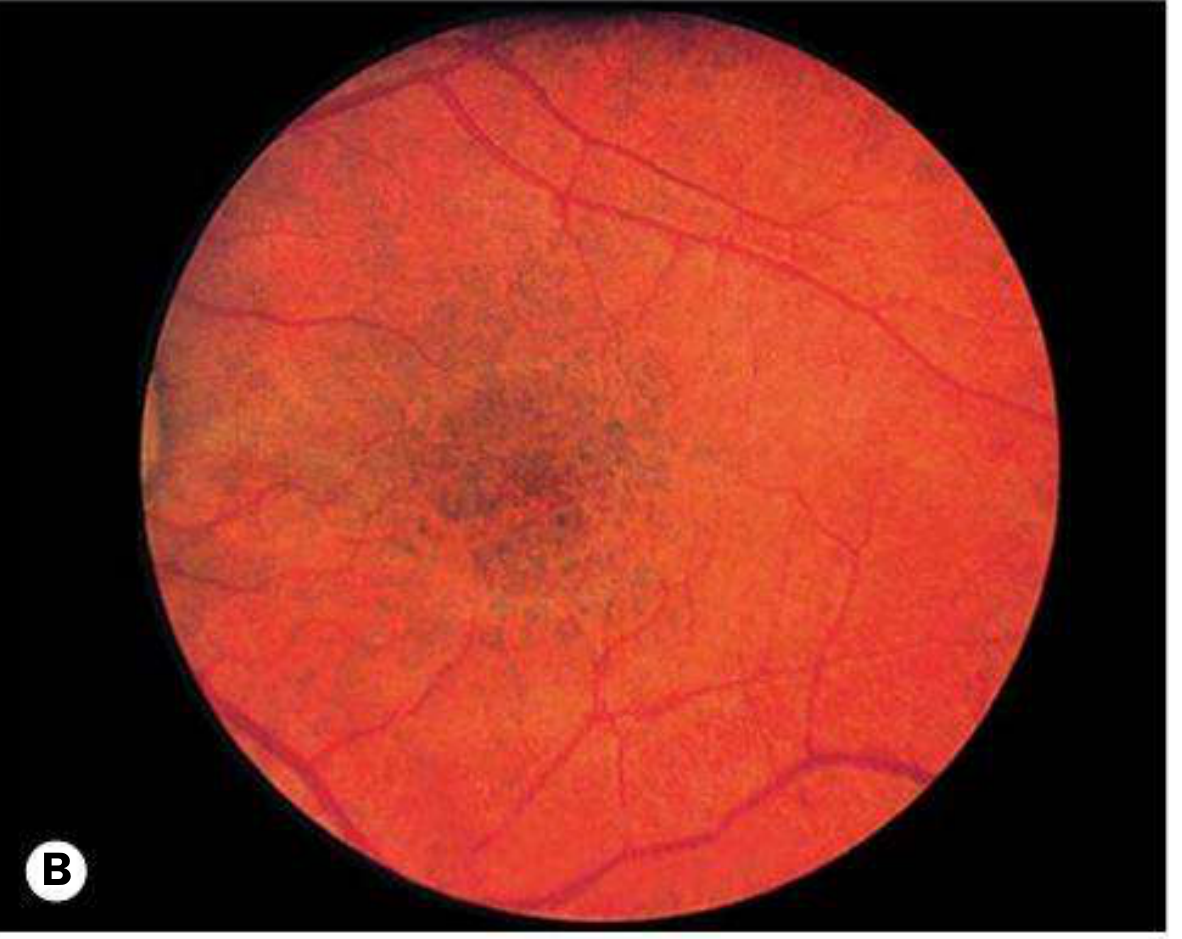
| Choroidal hyper-reflective nodules | >80% on OCT - multiple small flat pigmented lesions; highly sensitive and specific for NF1 |
| Choroidal naevi | Present; NF1 patients with naevi have increased risk of choroidal melanoma |
| Retinal 'corkscrew' vessels | ~1/3 of patients; do not leak |
| Congenital hypertrophy of RPE | May be more common than in unaffected individuals |
| Myelinated nerve fibres | Possibly increased |
| Combined hamartoma of retina and RPE | Possibly increased (especially in NF2) |
| Retinal astrocytic hamartoma | Identical to those seen in tuberous sclerosis |
Neurofibromatosis Type 2 (NF2)
Genetics: Autosomal dominant; NF2 gene on chromosome 22 (encodes Merlin protein); ~50% sporadic. Less common (1:40,000) but greater morbidity than NF1.
Key point: Ocular lesions are often the first manifestations of NF2, before the characteristic vestibular schwannomas become symptomatic.
Diagnostic Criteria (NF2) - Ocular Relevance
Posterior subcapsular lenticular opacities (juvenile cataract) are one of the characteristic lesions used in NF2 diagnostic criteria. From Goldman-Cecil Medicine:
- Bilateral vestibular schwannomas, OR
- Family history of NF2 + any 2 of: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities
Lens
- Juvenile posterior subcapsular/capsular cataract - affects ~60-80% of NF2 patients. Opacities develop before age 30. May be posterior subcapsular or capsular, cortical, or mixed. This is a cardinal diagnostic feature.
Fundus
- Epiretinal membrane - frequent
- Combined hamartoma of the retina and retinal pigment epithelium - relatively common
- Abnormal electroretinogram
Other Ophthalmic Features of NF2
- Ocular motor defects (~10%)
- Optic nerve sheath meningioma (less common) - children may present with this before an 8th nerve tumour
- Optic nerve glioma (less common)
- Unilateral Lisch nodules (uncommon; bilateral Lisch nodules point to NF1)
Summary Comparison Table
| Feature | NF1 | NF2 |
|---|---|---|
| Lisch nodules | ≥95% (bilateral, pathognomonic) | Rare/unilateral |
| Optic nerve glioma | 15-40% | Less common |
| Eyelid plexiform neurofibroma | Yes (S-shaped deformity) | No |
| Cataract | Uncommon | 60-80% (juvenile PSC) |
| Choroidal hyper-reflective nodules (OCT) | >80%, highly specific | Not a feature |
| Epiretinal membrane | No | Frequent |
| Combined hamartoma retina/RPE | Rare | Relatively common |
| Pulsating proptosis | Yes (sphenoid dysplasia) | No |
| Congenital glaucoma | ~50% with upper lid neurofibroma | Not typical |
| Ocular motor defects | Rare | ~10% |
Clinical Importance
- Regular eye examinations from the time of NF1 diagnosis are mandatory, primarily to detect optic nerve glioma early.
- In NF2, an ophthalmologist may be the first to diagnose the condition via juvenile cataract or combined retinal hamartoma.
- Choroidal OCT nodules are now recognized as a highly sensitive screening tool for NF1 (>80% sensitivity).
Sources: Kanski's Clinical Ophthalmology: A Systematic Approach, 10th ed., pp. 848-849; Goldman-Cecil Medicine, International Ed.; Fitzpatrick's Dermatology, Vol. 1-2
Give essay
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have all the depth needed for a full essay. Here it is:
Ocular Manifestations of Neurofibromatosis
Introduction
Neurofibromatosis (NF) encompasses a group of autosomal dominant neurocutaneous disorders - the phakomatoses - in which tumours and hamartomas develop along neural tissues and affect multiple organ systems, including the eye. The two clinically and genetically distinct forms most relevant to ophthalmology are Neurofibromatosis Type 1 (NF1, von Recklinghausen disease) and Neurofibromatosis Type 2 (NF2). Both carry characteristic and diagnostically important ocular findings; indeed, in NF2, ocular lesions are frequently the first manifestations of the disease. A thorough understanding of the ophthalmic features of both subtypes is essential not only for the early diagnosis of these conditions but also for preventing the serious visual morbidity that can result if they are left undetected.
Part I: Neurofibromatosis Type 1
Background and Genetics
NF1 is the most common phakomatosis, affecting approximately 1 in 3,000-4,000 individuals. It follows an autosomal dominant inheritance pattern with variable expressivity and irregular penetrance, though nearly 50% of cases arise from new (de novo) mutations. The causative gene, NF1, is located on chromosome 17q11.2 and encodes neurofibromin, a tumour suppressor protein that downregulates the RAS-MAPK signalling pathway by converting active RAS-GTP to inactive RAS-GDP. Loss of neurofibromin function leads to constitutive RAS activation, resulting in dysregulated cell growth and a predisposition to tumour formation throughout neural tissues. There is no consistent genotype-phenotype correlation.
The diagnosis of NF1 requires two or more of the following NIH criteria: (1) six or more café-au-lait macules (>5 mm prepuberty, >15 mm postpuberty); (2) two or more neurofibromas of any type or one plexiform neurofibroma; (3) axillary or inguinal freckling; (4) sphenoid bone dysplasia; (5) optic nerve glioma; (6) two or more Lisch nodules; and (7) a first-degree relative with NF1. Optic glioma and Lisch nodules are the two primary ophthalmic diagnostic criteria, underscoring how central the eye is to this diagnosis.
1. Eyelid: Plexiform Neurofibroma
The most clinically striking adnexal manifestation of NF1 is the plexiform neurofibroma of the upper eyelid. This lesion infiltrates and thickens the lid diffusely, producing the classic S-shaped deformity of the upper eyelid margin - a configuration considered virtually pathognomonic of NF1. On palpation, the tissue has a characteristic feel described as a "bag of worms," reflecting the tortuous enlargement of individual nerve fascicles within it. It may be associated with ptosis, amblyopia in children, and facial hemiatrophy.
In approximately 50% of cases where congenital glaucoma develops in NF1, there is an ipsilateral plexiform neurofibroma of the upper eyelid and facial hemiatrophy. The proposed mechanism involves either direct infiltration of the anterior angle or a developmental anomaly of trabecular meshwork induced by the neurofibroma. Mechanical ptosis from eyelid infiltration also risks deprivation amblyopia in young children, making early ophthalmological assessment mandatory.
2. Orbit
Optic Nerve Glioma
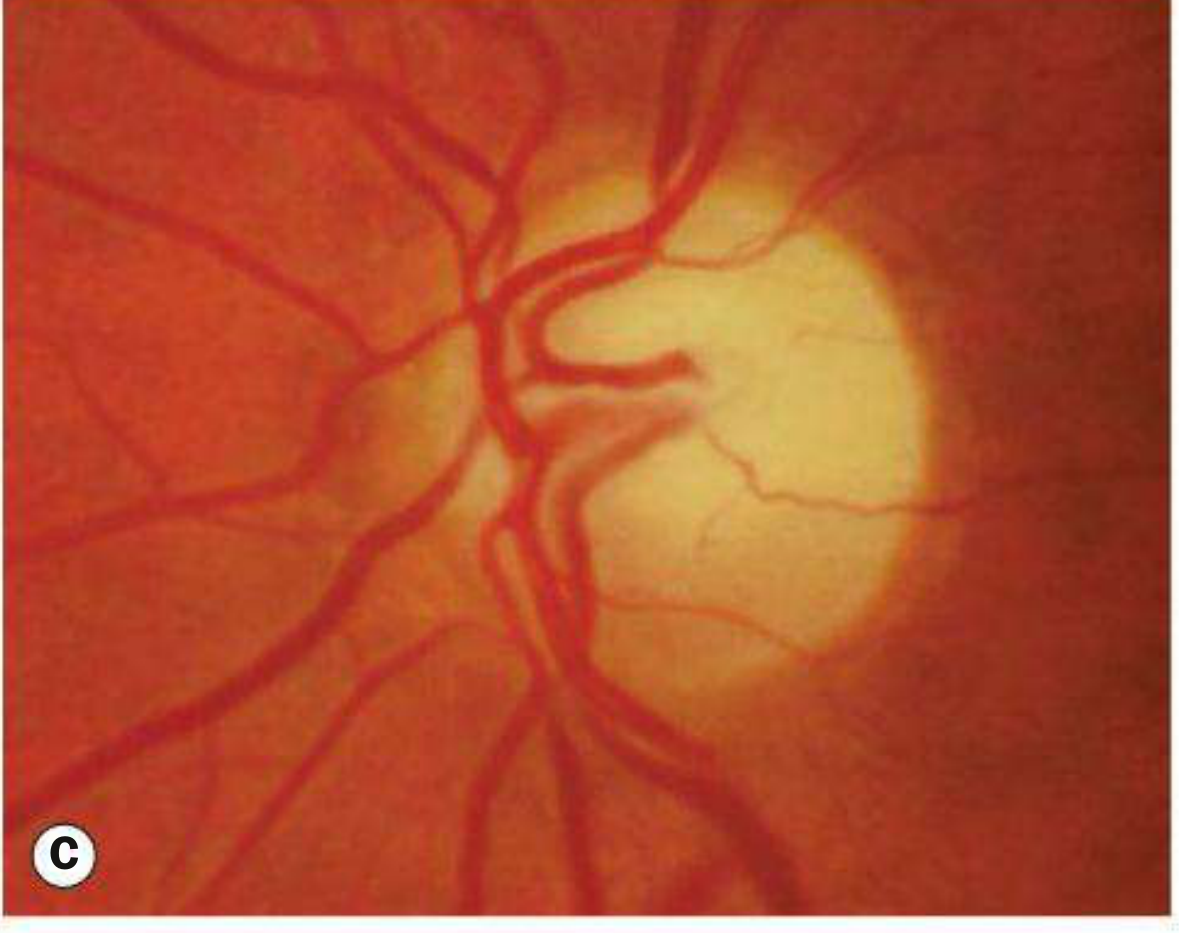
The most important orbital and indeed overall ophthalmic complication of NF1 is optic nerve glioma, occurring in approximately 15-40% of patients. Histologically, it is a pilocytic astrocytoma - a low-grade (WHO grade 1) tumour composed of spindle-shaped pilocytic (hair-like) astrocytes and glial filaments. It typically presents in young children, with a median age of onset around 6.5 years, and produces a fusiform enlargement of the optic nerve.
Clinical presentation is with slowly progressive, painless proptosis (often non-axial, with inferior or temporal dystopia), marked visual impairment, optic atrophy, and strabismus. Bilateral involvement is possible, and the tumour may extend posteriorly along the visual pathway to involve the optic chiasm, optic tract, and hypothalamus, occasionally causing obstructive hydrocephalus. Chiasmatic involvement in children with NF1 is sometimes heralded by precocious puberty. Paradoxically, optic gliomas in NF1 generally behave less aggressively and have a better prognosis than sporadic optic gliomas in non-NF1 patients.
Management options include observation with serial MRI, carboplatin/vincristine-based chemotherapy, temozolomide, and more recently the MEK inhibitor selumetinib (25 mg/m² twice daily), which has demonstrated the ability to reduce the size of inoperable tumours in clinical trials. Radiation is avoided in NF1 patients due to the risk of secondary malignancies and moyamoya vasculopathy.
Spheno-orbital Encephalocoele
Absence or dysplasia of the greater wing of the sphenoid bone is a characteristic skeletal abnormality of NF1. In the orbit, this can permit herniation of brain parenchyma into the orbit, creating a pulsating proptosis - a distinctive and diagnostically important sign. The pulsations arise because the orbital contents receive transmitted pressure waves from cerebrospinal fluid and cerebral arterial pulsations in the absence of the sphenoid bony wall.
Other Orbital Neural Tumours
Less commonly, other neural tumours may develop in the orbit in NF1, including neurilemmomas (schwannomas), additional plexiform neurofibromas, and meningiomas.
3. Iris Lesions
Lisch Nodules
Lisch nodules are the most pathognomonic ocular sign of NF1 and are present in at least 95% of adult patients. They are melanocytic hamartomas of the iris stroma, appearing as tiny dome-shaped, well-circumscribed, pigmented lesions that protrude above the iris surface. They are best seen on slit-lamp examination. They develop progressively during the second and third decades of life and are bilateral by definition.
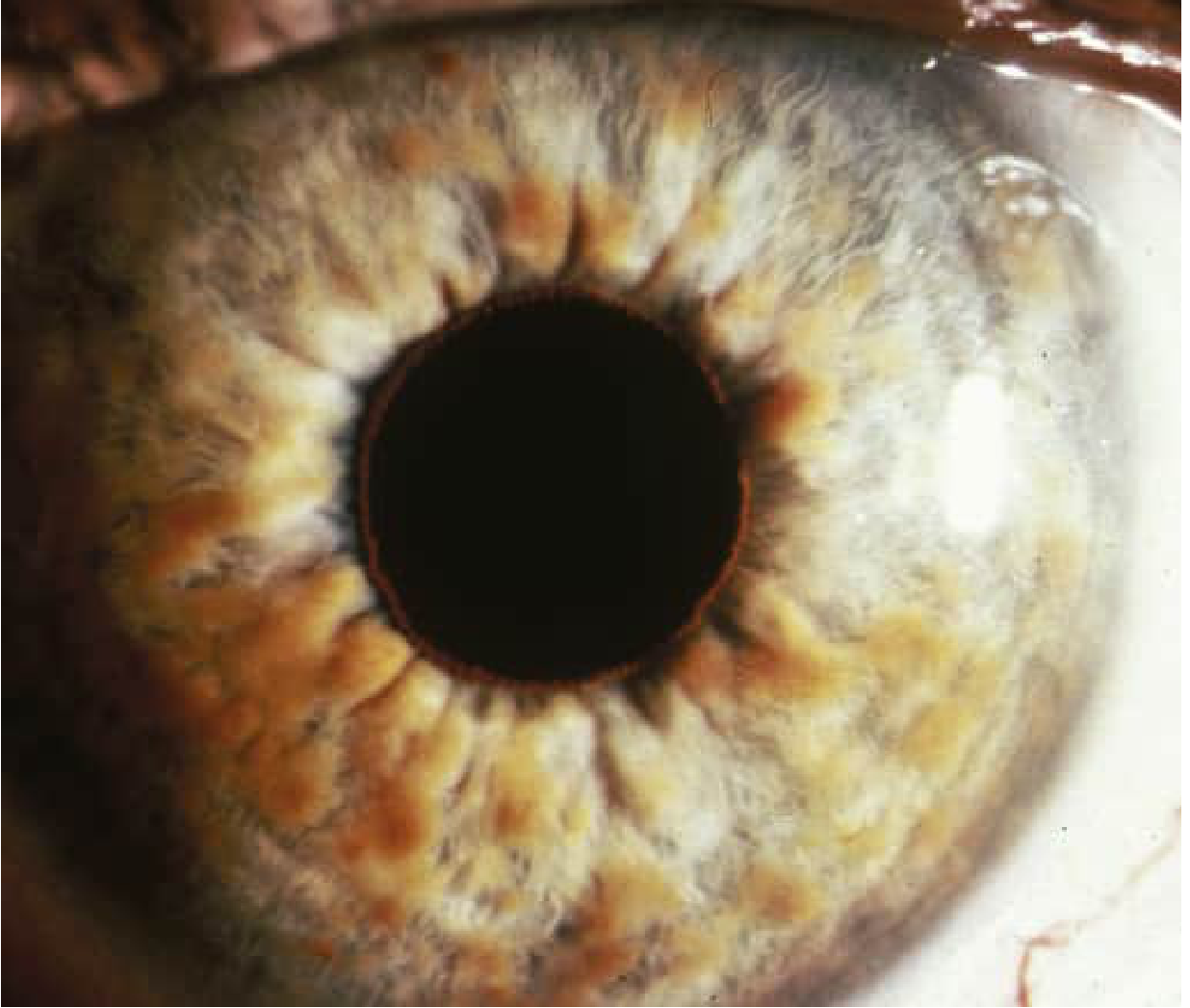
Lisch nodules do not affect vision and carry no intrinsic clinical consequences, but their presence is enormously valuable diagnostically, particularly in children under 6 years of age in whom café-au-lait spots are the dominant early finding. Finding bilateral Lisch nodules effectively confirms a clinical diagnosis of NF1 when combined with other criteria.
Congenital Ectropion Uveae
Congenital ectropion uveae - a migration of posterior iris pigment epithelium onto the anterior iris surface - may occur in NF1, though it is uncommon. It may be associated with glaucoma and should prompt assessment of intraocular pressure and angle anatomy.
Mammillations
Diffuse iris mammillations (innumerable tiny, uniform nodular surface elevations imparting a cobblestone texture to the iris) are a rare finding in NF1, distinct from Lisch nodules.
4. Cornea
Prominent corneal nerves may be observed in NF1, appearing as thickened, visible nerve trunks in the corneal stroma on slit-lamp biomicroscopy. While not specific to NF1 (they also occur in MEN 2B, Fuchs corneal dystrophy, and other conditions), their presence in the context of other findings supports the diagnosis.
5. Glaucoma
Glaucoma is not a frequent association in NF1 overall. When it does occur, it is typically unilateral and congenital (developmental glaucoma). As noted, approximately half of NF1 patients who develop congenital glaucoma also have an ipsilateral plexiform neurofibroma of the upper eyelid and ipsilateral facial hemiatrophy. The mechanism may involve angle dysgenesis secondary to neural crest abnormalities, or direct infiltration of angle structures by neurofibroma tissue.
6. Fundus and Choroidal Findings
Choroidal Hyper-reflective Nodules
Perhaps the most diagnostically significant posterior segment finding in NF1 in modern practice is the presence of choroidal hyper-reflective nodules visible on optical coherence tomography (OCT). These are multiple small, flat, pigmented lesions within the choroid that are not readily visible on conventional fundoscopy but are consistently detected with infrared reflectance imaging and OCT. They are found in over 80% of NF1 patients and are considered highly sensitive and specific for the condition. Their detection has become an important screening and diagnostic adjunct.
Choroidal Naevi
Choroidal naevi may be found in NF1 patients. Importantly, NF1 patients with choroidal naevi have an increased risk of developing choroidal melanoma compared to the general population, a consequence of the same tumour suppressor dysfunction that underlies other neoplastic tendencies in NF1.
Retinal 'Corkscrew' Vessels
Tortuous, 'corkscrew'-shaped retinal vessels are found in approximately one third of NF1 patients. They do not leak on fluorescein angiography and are thought to represent a vascular dysplasia rather than a secondary ischaemic change.
Other Fundus Findings
Additional posterior segment findings that may be seen with increased frequency in NF1 include congenital hypertrophy of the retinal pigment epithelium (CHRPE), myelinated nerve fibres, combined hamartoma of the retina and RPE, and retinal astrocytic hamartoma (identical to those seen in tuberous sclerosis). These are individually uncommon but collectively form part of the broad ocular phenotype.
Part II: Neurofibromatosis Type 2
Background and Genetics
NF2 is far less common than NF1, with an estimated birth incidence of 1 in 40,000-50,000 live births. It is also autosomal dominant, but with 50% of cases arising de novo. The NF2 gene resides on chromosome 22q12 and encodes merlin (also called schwannomin), a cytoskeletal protein related to the ERM (ezrin-radixin-moesin) family that normally acts as a tumour suppressor by regulating contact-dependent inhibition of cell growth. Loss of merlin leads to uncontrolled Schwann cell proliferation, explaining the predilection for schwannomas. Despite being categorised together with NF1, NF2 bears little phenotypic resemblance to it - there are few café-au-lait spots, no Lisch nodules (as a rule), and no neurofibromas of the plexiform type.
The hallmark systemic feature is bilateral vestibular (8th cranial nerve) schwannomas, which cause progressive sensorineural hearing loss, tinnitus, and vertigo, typically becoming symptomatic in young adulthood. Approximately 60% of patients present in adulthood with hearing loss. Patients also develop multiple meningiomas, spinal ependymomas, and peripheral schwannomas. The morbidity of NF2 is substantially greater than NF1, with many patients ultimately becoming paralysed and deaf.
A critically important clinical point: ocular lesions in NF2 are frequently the first manifestations of the disease, preceding the development of symptomatic vestibular schwannomas, sometimes by years. Children with NF2 are more likely to present with ocular or non-eighth-nerve tumour manifestations than adults. The ophthalmologist therefore plays a pivotal role in early diagnosis.
1. Lens - Juvenile Posterior Subcapsular Cataract
The most common and diagnostically important ocular finding in NF2 is juvenile cataract, affecting 60-80% of patients. The opacities develop before age 30, distinguishing them from age-related cataract, and may be posterior subcapsular, capsular, cortical, or a mixture of types. Posterior subcapsular lens opacities are included as one of the characteristic lesions in the NF2 diagnostic criteria - a young patient found to have posterior subcapsular cataract with a family history of NF2 is presumed to have the condition until proven otherwise. The mechanism is thought to relate to merlin's role in epithelial cell proliferation; loss of its function may allow aberrant migration of lens epithelial cells to the posterior pole.
2. Fundus - Combined Hamartoma of the Retina and RPE
Combined hamartoma of the retina and retinal pigment epithelium (CHRRPE) is relatively common in NF2 and may be the presenting finding. It appears as a slightly elevated, pigmented lesion with overlying retinal distortion, abnormal vessels, and epiretinal membrane formation. It typically causes mild visual disturbance or is found incidentally. CHRRPE is considered the most specific fundus lesion for NF2.
Epiretinal Membrane
Epiretinal membrane is a frequent finding in NF2, and may be the substrate producing the tractional distortion seen in CHRRPE. It may also occur in isolation, causing visual distortion (metamorphopsia).
3. Optic Nerve and Visual Pathway
Optic Nerve Sheath Meningioma
Although less common than cataract, optic nerve sheath meningioma occurs in NF2 and is a particularly important lesion in children, who are more likely to present with this tumour than with the classical vestibular schwannoma. It presents with slowly progressive visual loss, proptosis, and optic disc oedema followed by atrophy, with the characteristic "railroad track" or "tram-track" sign on CT or MRI (enhancement of the periphery of the optic nerve sheath with a central lucent stripe).
Optic Nerve Glioma
Optic nerve glioma can also occur in NF2, though it is far less common than in NF1.
4. Ocular Motility Defects
Cranial nerve palsies affecting extraocular movement occur in approximately 10% of NF2 patients, reflecting the involvement of cranial nerves by schwannomas or meningiomas along the skull base.
5. Lisch Nodules in NF2
Bilateral Lisch nodules, which are the hallmark iris finding of NF1, are absent or rare in NF2. Occasional unilateral Lisch nodules have been reported, but their presence should always raise the question of NF1 rather than NF2.
6. Electroretinogram Abnormalities
An abnormal electroretinogram has been recorded in NF2 patients, reflecting diffuse retinal dysfunction beyond what would be expected from focal lesions alone.
Comparative Summary
| Feature | NF1 | NF2 |
|---|---|---|
| Gene / Chromosome | NF1 / Chr 17 | NF2 / Chr 22 |
| Protein | Neurofibromin | Merlin |
| Prevalence | 1:3,000-4,000 | 1:40,000-50,000 |
| Lisch nodules | ≥95%, bilateral, pathognomonic | Absent / rare |
| Optic nerve glioma | 15-40% | Uncommon |
| Plexiform neurofibroma (eyelid) | Yes - S-shaped lid deformity | No |
| Juvenile cataract | Uncommon | 60-80%, PSC type |
| Choroidal OCT nodules | >80%, highly specific | Not a feature |
| Epiretinal membrane | Rare | Frequent |
| Combined hamartoma retina/RPE | Uncommon | Relatively common |
| Optic nerve sheath meningioma | Less common | NF2-associated (especially in children) |
| Pulsating proptosis | Yes (sphenoid dysplasia) | No |
| Congenital glaucoma | Yes (with upper lid neurofibroma) | Not typical |
| Ocular motor defects | Rare | ~10% |
| First ocular presentation | Not typical | Frequent (cataract, CHRRPE) |
Management Implications
Both NF1 and NF2 require regular ophthalmic surveillance from the time of diagnosis. In NF1, the primary goal of surveillance is the early detection of optic pathway glioma, for which serial MRI and visual function testing are used; visual evoked potentials and colour vision assessment help detect subclinical involvement. Management of inoperable plexiform neurofibromas may include the MEK inhibitor selumetinib. Congenital glaucoma requires early surgical intervention (goniotomy or trabeculotomy). Amblyopia from eyelid ptosis needs urgent patching therapy.
In NF2, ophthalmological examination is valuable both for establishing an early diagnosis (via juvenile cataract or CHRRPE) and for ongoing surveillance of visual pathway involvement. Combined hamartomas do not typically require treatment unless vision is significantly affected. Optic nerve sheath meningiomas may require stereotactic radiosurgery. Cataract extraction should be performed when visually significant.
Conclusion
The eye is a window to neurofibromatosis. In NF1, the finding of bilateral Lisch nodules on slit-lamp examination is pathognomonic and forms a formal diagnostic criterion; optic nerve glioma is the leading cause of visual morbidity and requires lifelong surveillance. In NF2, juvenile posterior subcapsular cataract and combined retinal hamartoma may precede the development of vestibular schwannomas by years and allow earlier diagnosis and counselling. Across both types, the ophthalmologist occupies a central position in the diagnosis, monitoring, and management of these complex multisystem conditions.
Sources: Kanski's Clinical Ophthalmology: A Systematic Approach, 10th ed., pp. 848-849; Goldman-Cecil Medicine, International Ed., Chs. 209 & 385; Bradley and Daroff's Neurology in Clinical Practice, Box 99.2; Fitzpatrick's Dermatology, Vol. 1-2, Ch. 135
This is a shared conversation. Sign in to Orris to start your own chat.