Isoniazid medicinal chemistry
Reading File
Finding Sources
Searching the Web
isoniazid medicinal chemistry structure mechanism SAR
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Loading Image
Reading File
Reading File
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full medicinal chemistry answer:
Isoniazid (INH) - Medicinal Chemistry
1. Chemical Structure & Physical Properties
Isoniazid (isonicotinic acid hydrazide, INH) is a remarkably simple synthetic molecule:
- Chemical name: Isonicotinic acid hydrazide
- Molecular formula: C₆H₇N₃O
- Molecular weight: 137.14 Da
- Structure: A pyridine ring (at the 4-position) bearing a hydrazide (-CO-NH-NH₂) functional group
- Physical form: Colourless crystalline solid; water-soluble (highly hydrophilic for a drug of its size)
- Synthesis: Manufactured from 4-cyanopyridine + hydrazine hydrate; alternatively from citric acid. First synthesised in 1912 by Meyer and Mally at Charles University, Prague - its antitubercular activity was only discovered 40 years later in 1952.
The structure contains two key pharmacophoric elements:
- The pyridine ring - provides the nicotinoyl scaffold required for NAD adduct formation
- The hydrazide (-CO-NHNH₂) group - the site of KatG-mediated activation; essential for prodrug bioactivation
2. Mechanism of Action - Prodrug Activation
Isoniazid is a prodrug - it has no direct antimycobacterial activity and must be bioactivated inside the mycobacterium.
Step 1: Passive diffusion
INH enters mycobacterial cells by passive diffusion (small, uncharged molecule).
Step 2: KatG activation
Inside the bacterium, KatG (a multifunctional mycobacterial catalase-peroxidase encoded by the katG gene) oxidises isoniazid, generating a cascade of reactive species:
- Isonicotinoyl radical (most critical)
- Acyl peroxy radicals
- Pyridyl radicals
- Reactive oxygen species: superoxide, H₂O₂, alkyl hydroperoxides
- Nitric oxide (NO) radical - contributes to mycobactericidal effect
Step 3: NAD Adduct formation
The isonicotinoyl radical spontaneously couples with NADH (reduced nicotinamide adenine dinucleotide) to form the isonicotinoyl-NAD adduct - the true active species.
A distinct nicotinoyl-NADP isomer adduct also forms and potently inhibits mycobacterial dihydrofolate reductase (Ki < 1 nM), interfering with nucleic acid synthesis.
Step 4: Target inhibition - Mycolic Acid Synthesis
The isonicotinoyl-NAD complex binds tightly to two key enzymes in the mycobacterial fatty acid synthesis type II (FAS-II) system:
| Target Enzyme | Gene | Role |
|---|---|---|
| InhA (enoyl-acyl carrier protein reductase; NADH-dependent) | inhA | Reduces long-chain enoyl fatty acids in mycolic acid elongation |
| KasA (β-ketoacyl-ACP synthase) | kasA | Condenses fatty acid units; involved in mycolic acid elongation |
Blocking InhA and KasA halts synthesis of mycolic acids - long-chain (C60-C90) hydroxylated fatty acids unique to mycobacteria. These are absolutely essential components of the mycobacterial cell wall (the outer lipid layer / "mycomembrane"), and their loss causes fatal disruption of cell wall integrity.
Why selectivity? Mycolic acids are unique to mycobacteria. Human cells do not have FAS-II or InhA homologs that INH targets, explaining high selectivity and low host toxicity at therapeutic doses.
3. Antibacterial Spectrum
- Highly specific for M. tuberculosis (MIC 0.025-0.1 μg/mL for wild-type strains)
- Active against M. kansasii at higher concentrations (MIC 0.5-2 μg/mL)
- Moderate activity against M. bovis
- Poor activity against MAC (Mycobacterium avium complex)
- No activity against any non-mycobacterial genus
- Particularly effective against rapidly dividing bacilli; also active against intracellular organisms within macrophages
Sources: Goodman & Gilman's, Harrison's Principles of Internal Medicine 22E, Katzung Basic & Clinical Pharmacology 16E
4. Pharmacokinetics
| Parameter | Detail |
|---|---|
| Administration | Oral (tablets) or IM |
| Absorption | Rapid; food (especially high-fat meals) reduces Cmax by up to 50%; antacids reduce absorption |
| Typical oral dose | 300 mg/day in adults (5 mg/kg/d) |
| Peak plasma level | 3-5 μg/mL within 30 min to 2 hours |
| Distribution | Excellent - penetrates all body fluids, cells, caseous material (necrotic TB lesions), CSF (concentrations equal to serum) |
| Protein binding | Low |
| Metabolism | Hepatic: N-acetylation (NAT2) + hydrolysis → inactive metabolites |
| Excretion | Renal (glomerular filtration + tubular secretion), predominantly as metabolites |
Pharmacogenomic Acetylation Polymorphism (NAT2)
This is one of the classic examples of pharmacogenomics in medicine. Isoniazid is acetylated by N-acetyltransferase 2 (NAT2) - a polymorphic enzyme with a bimodal population distribution:
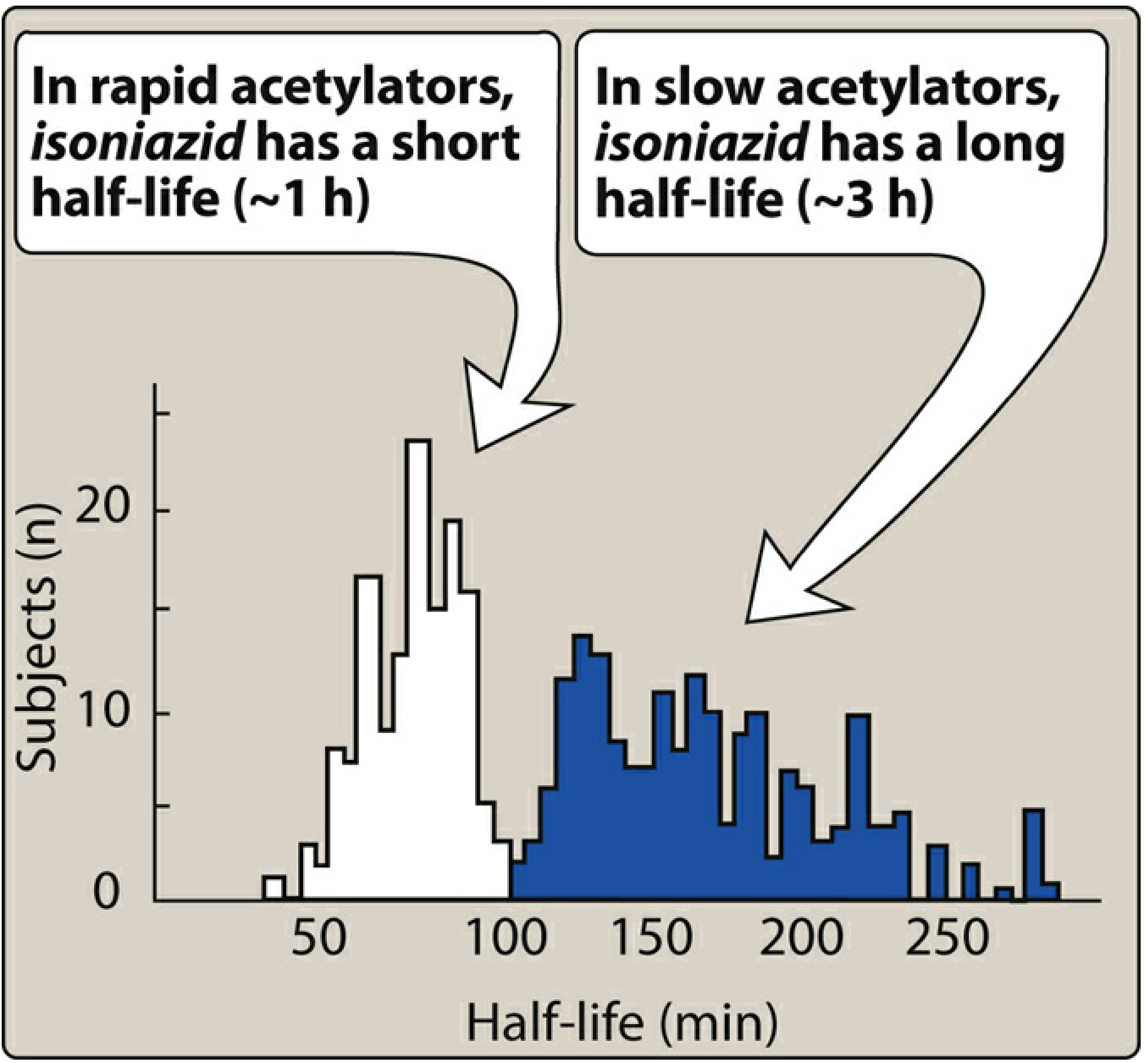
Figure: Bimodal half-life distribution - rapid acetylators ~1 hour (90 min), slow acetylators ~3-4 hours (Lippincott Illustrated Reviews: Pharmacology)
| Phenotype | Half-life | Average plasma conc. | Clinical implication |
|---|---|---|---|
| Rapid acetylators | ~90 min (< 1 h) | 1/3 to 1/2 of slow | Subtherapeutic levels with weekly dosing or malabsorption; higher risk of hepatotoxicity via toxic acetyl metabolites |
| Slow acetylators | 3-4 hours | Higher | More parent drug excreted; higher risk of peripheral neuropathy |
Slow acetylators excrete more unchanged parent compound. Genotyping via NAT2 is increasingly used to characterise pharmacogenomic responses.
5. Resistance Mechanisms
Resistance arises from chromosomal mutations (frequency ~1 in 10⁵ bacilli):
| Mechanism | Gene | Resistance Level | Notes |
|---|---|---|---|
| Loss/mutation of activating enzyme | katG | High-level | Most common (~70% of resistant isolates). Key mutation: Ser315Asn in heme-binding domain - loses ability to form NAD adducts but retains catalase activity and biofitness |
| Overexpression of target enzyme | inhA promoter | Low-level | Reduces binding affinity; also confers cross-resistance to ethionamide |
| Overexpression of ahpC (alkyl hydroperoxide reductase) | ahpC promoter | Low-level | Detoxifies organic peroxides generated by KatG; compensatory mutation in katG-mutant strains |
| Mutations in fatty acid synthase | kasA | Variable | Less common |
| Loss of NADH dehydrogenase 2 activity | ndh | Variable | Confers INH resistance |
Because resistant mutants pre-exist at ~1/10⁵, and TB cavities contain 10⁷-10⁹ organisms, monotherapy inevitably selects for resistance. Two drugs together give a probability of dual resistance of ~1/10¹² - effectively negligible.
Sources: Goodman & Gilman's, Katzung 16E, Harrison's 22E
6. Drug Interactions
INH is a CYP450 inhibitor (multiple isoforms):
- Increases levels of: phenytoin, carbamazepine, benzodiazepines, warfarin
- When co-administered with rifampin (a potent CYP inducer), the net effect usually reduces the levels of these co-medications
7. Adverse Effects - Mechanistic Basis
| Adverse Effect | Mechanism | Risk Factors |
|---|---|---|
| Hepatotoxicity (most serious) | Acetyl hydrazine metabolite (from N-acetylation of INH) → covalent binding to liver proteins → hepatocellular necrosis; possible immune mechanism | Age >50 yr, alcohol use, pre-existing liver disease, rapid acetylators (more acetyl hydrazine) |
| Peripheral neuropathy | INH promotes excretion of pyridoxine (B₆); pyridoxal-5'-phosphate (P5P) is a cofactor for many enzymes including those in GABA synthesis | Slow acetylators (more parent drug), malnutrition, diabetes, alcohol use, AIDS, renal disease |
| CNS toxicity (seizures, psychosis, ataxia) | P5P depletion prevents glutamate decarboxylase from making GABA (the main inhibitory neurotransmitter). Classic toxicity in overdose | Overdose, pyridoxine deficiency |
| Drug-induced lupus | Immune mechanism | - |
| Haematologic | Pyridoxine-deficiency anaemia, sideroblastic anaemia | Slow acetylators |
Antidote for overdose: Pyridoxine (1 g per gram of INH ingested) - replenishes P5P, restores GABA synthesis.
Prevention: Co-administration of pyridoxine 25-50 mg/day prevents peripheral neuropathy in high-risk patients.
8. Structure-Activity Relationships (SAR)
| Structural Feature | Role | Effect of Modification |
|---|---|---|
| Pyridine ring (isonicotinoyl) | Required for NAD adduct formation; the ring nitrogen participates in adduct geometry | Replacement abolishes activity |
| Hydrazide group (-CO-NHNH₂) | Site of KatG oxidation (prodrug activation) | Essential; replacing with ester → loss of activity |
| Free NH₂ of hydrazide | Needed for KatG recognition and radical formation | Substitution reduces or abolishes activity |
| Position 4 substitution of pyridine | Optimal positioning for InhA binding of the NAD adduct | Position 3 (nicotinoyl hydrazide) much less active |
| Acyl hydrazide vs simple hydrazine | Carbonyl adjacent to hydrazide important for KatG activation | Simple pyridyl hydrazine is far less active |
Ethionamide (a thioamide) shares partial mechanistic overlap with INH at InhA (it also forms an InhA-NAD adduct via its own activation), explaining cross-resistance when inhA is overexpressed.
Summary
Isoniazid is a deceptively simple yet highly sophisticated antimycobacterial prodrug. Its pyridine-hydrazide scaffold is chemically unremarkable, but the drug's entire pharmacology depends on its bioactivation by the mycobacterium's own enzyme (KatG) to generate an isonicotinoyl-NAD adduct that blocks InhA - a target exquisitely unique to mycobacterial fatty acid metabolism. This selectivity, combined with excellent tissue penetration (including CSF and caseous lesions), makes INH the backbone of TB chemotherapy. Its main pharmacogenomic feature - NAT2 polymorphism - shapes both its toxicity profile and dosing considerations across populations.
Sources:
- Goodman & Gilman's Pharmacological Basis of Therapeutics
- Harrison's Principles of Internal Medicine, 22nd Edition (2025)
- Katzung Basic and Clinical Pharmacology, 16th Edition
- Lippincott Illustrated Reviews: Pharmacology
- Frontiers in Pharmacology (2024) - INH historical development, metabolism & SAR
This is a shared conversation. Sign in to Orris to start your own chat.