Explain apoptosis
Reading File
Searching the Web
apoptosis mechanisms pathways intrinsic extrinsic 2024
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive textbook content from Robbins and Sleisenger, plus two excellent diagrams. Here is the full, detailed explanation:
Apoptosis
Apoptosis is a pathway of programmed cell death in which cells activate enzymes that degrade their own nuclear DNA and cytoplasmic proteins. Unlike necrosis, the cell membrane stays intact throughout the process, cell contents do not leak out, and no inflammatory reaction is triggered. Fragments of the dying cell ("apoptotic bodies") are recognized and phagocytosed by macrophages before cellular contents can escape.
The word itself comes from the Greek for "falling off" - describing the way cells shed membrane-bound fragments as they die.
Morphological Features
Under a microscope, apoptotic cells show a recognizable sequence of changes (H&E staining):
- Cell shrinkage - the cytoplasm condenses
- Chromatin condensation - nuclear chromatin compacts against the nuclear envelope
- Nuclear fragmentation (karyorrhexis) - the nucleus breaks apart
- Cytoplasmic budding - membrane blebs form
- Apoptotic body formation - the cell fragments into membrane-bound packages containing intact organelles and nuclear fragments
- Phagocytosis - macrophages rapidly engulf these bodies without any inflammation
Because this process is so efficient, even widespread apoptosis can be histologically invisible.
The Two Main Pathways
Both pathways converge on the activation of caspases - a family of cysteine proteases that cleave proteins after aspartate residues. There are initiator caspases (8, 9) and executioner caspases (3, 7).
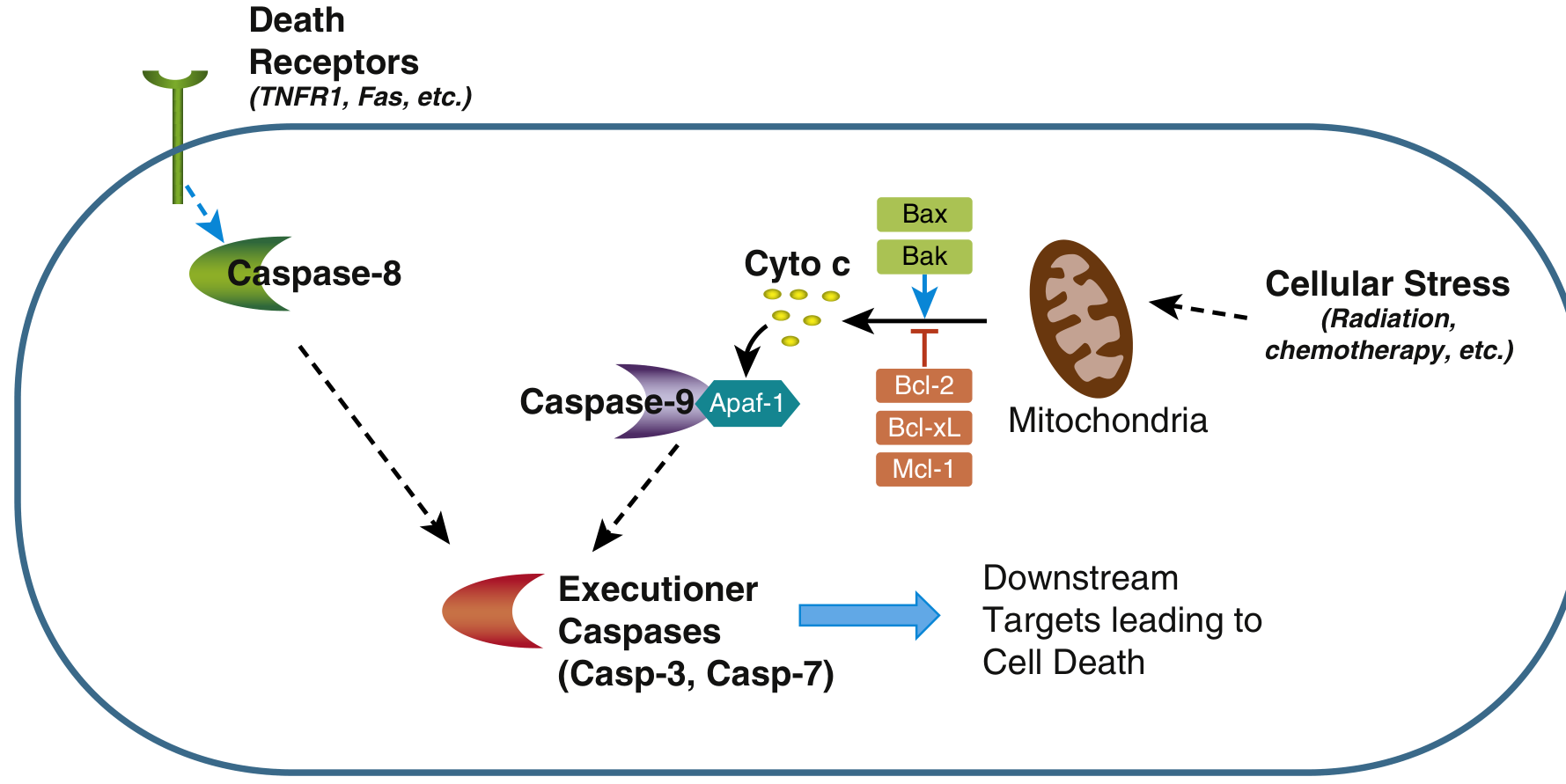
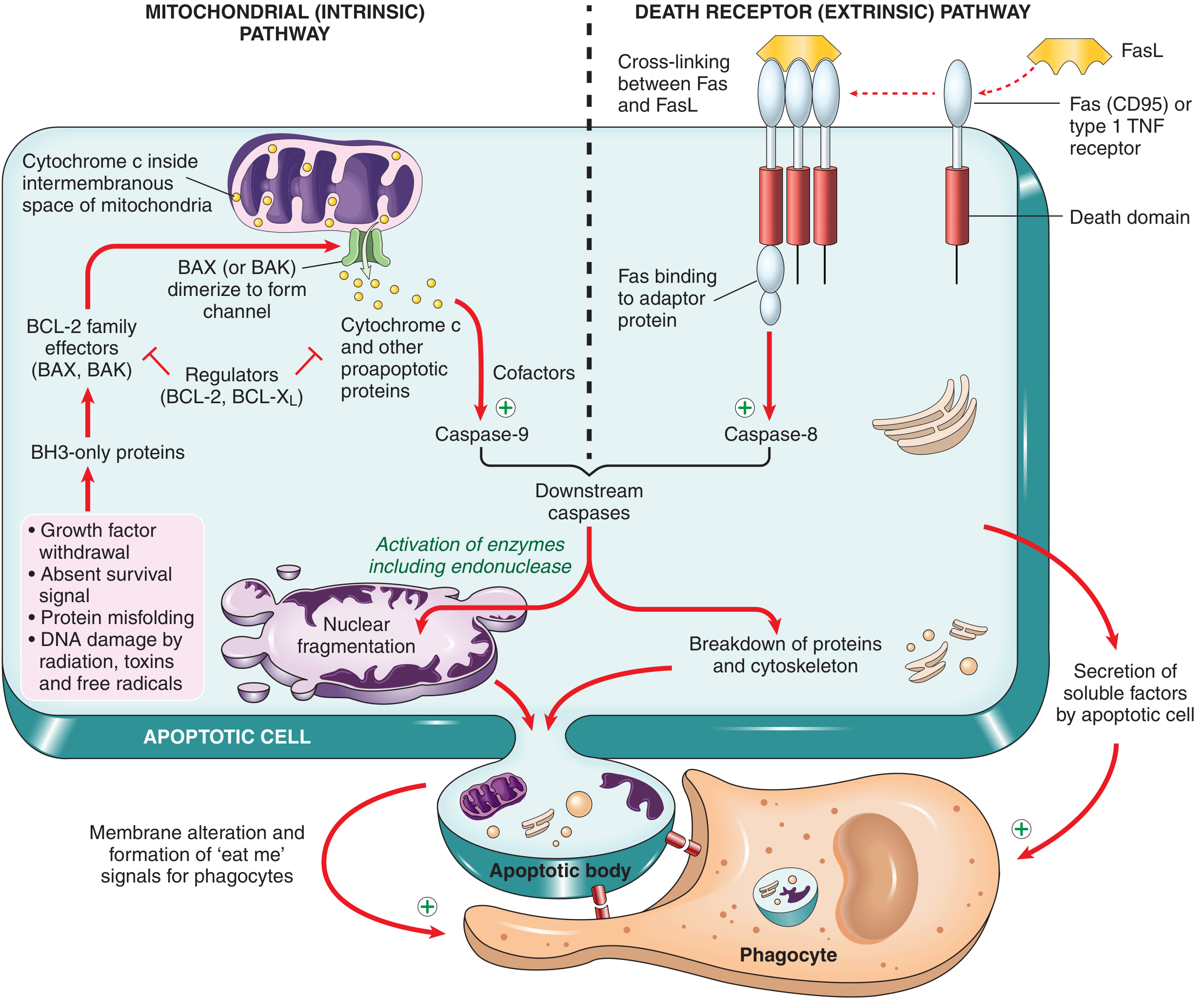
1. Mitochondrial (Intrinsic) Pathway
This is the dominant pathway in most physiologic and pathologic apoptosis. It is activated by signals arising inside the cell.
Triggers:
- Growth factor or survival signal withdrawal
- DNA damage (radiation, toxins, free radicals)
- Protein misfolding / ER stress
- Hypoxia or nutrient deprivation
Key steps:
- Cellular stress activates BH3-only proteins (sensors that belong to the BCL-2 family)
- BH3-only proteins shift the balance toward BAX and BAK (pro-apoptotic)
- BAX/BAK dimerize and insert into the outer mitochondrial membrane, forming channels
- Cytochrome c leaks out of the mitochondrial intermembrane space into the cytosol
- Cytochrome c + APAF-1 + caspase-9 precursors assemble into the apoptosome
- The apoptosome activates caspase-9 (initiator caspase)
- Caspase-9 activates executioner caspases 3 and 7
The BCL-2 family - the master regulators:
- Anti-apoptotic (inhibit pore formation): BCL-2, BCL-XL, MCL-1 - maintained by growth factor signaling
- Pro-apoptotic (form the pore): BAX, BAK
- Sensors (activate BAX/BAK): BH3-only proteins (e.g., BIM, PUMA, NOXA, BAD)
The ratio of anti- to pro-apoptotic BCL-2 family members ultimately determines whether a cell lives or dies.
2. Death Receptor (Extrinsic) Pathway
This pathway is activated by signals from outside the cell, via cell-surface death receptors.
Key receptors:
- Fas (CD95) - activated by Fas Ligand (FasL), expressed on activated T lymphocytes
- TNFR1 (type I TNF receptor) - activated by TNF-alpha
Key steps:
- FasL (on T cells) or TNF binds and cross-links death receptors on the target cell
- The receptor's cytoplasmic "death domain" recruits adaptor proteins (e.g., FADD)
- These adaptor proteins recruit and activate caspase-8 (initiator caspase)
- Active caspase-8 directly activates executioner caspases 3 and 7
Physiologic roles:
- Elimination of self-reactive lymphocytes (peripheral tolerance)
- CTL-mediated killing of virus-infected cells
- Termination of immune responses (lymphocyte contraction)
3. Execution Phase (Both Pathways Converge Here)
Activated caspases 3 and 7 cleave a large number of downstream substrates, driving the visible morphological changes:
| Substrate | Effect |
|---|---|
| CAD (caspase-activated DNase) inhibitor (ICAD) | Releases CAD, which fragments DNA into nucleosome-sized pieces (internucleosomal cleavage) |
| Nuclear lamins | Nuclear envelope collapse and fragmentation |
| Cytoskeletal proteins | Cell shrinkage and budding |
| Phosphatidylserine flippase | Exposes PS on the outer leaflet of the membrane ("eat me" signal) |
4. Clearance of Apoptotic Cells
Dying cells attract phagocytes via:
- Phosphatidylserine flipping to the outer membrane leaflet (recognized by macrophage receptors)
- Secreted chemotactic "find me" signals
Phagocytosis is so efficient that there is no inflammation - this is a defining feature distinguishing apoptosis from necrosis.
Physiologic vs. Pathologic Apoptosis
| Physiologic | Pathologic |
|---|---|
| Embryonic tissue sculpting (digit separation, neural tube closure) | Severe DNA damage (radiation, cytotoxic drugs) |
| Intestinal epithelial turnover | Accumulation of misfolded proteins (ER stress) |
| Involution of hormone-dependent tissues (endometrium) | Viral infections (some viruses hijack or induce apoptosis) |
| Lymphocyte homeostasis after immune responses | Ischemia-reperfusion injury |
| Deletion of self-reactive lymphocytes | Neurodegeneration |
Apoptosis vs. Necrosis
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Mechanism | Programmed, energy-dependent | Passive, energy-independent |
| Cell size | Shrinks | Swells |
| Membrane | Intact | Disrupted |
| Chromatin | Condenses, fragments neatly | Clumping, then dissolution |
| Contents | Contained in apoptotic bodies | Leaked into extracellular space |
| Inflammation | None | Yes (content leakage triggers it) |
| Caspases | Required | Not required |
Clinical Relevance
- Cancer: Mutations in BCL-2 (e.g., t(14;18) in follicular lymphoma), p53, or caspases allow cells to evade apoptosis. BCL-2 overexpression is one of the most common oncogenic mechanisms.
- Autoimmunity: Defective Fas/FasL signaling causes autoimmune lymphoproliferative syndrome (ALPS) - self-reactive lymphocytes fail to be deleted.
- Neurodegeneration: Excessive apoptosis contributes to neuron loss in Alzheimer's, Parkinson's, and ALS.
- Therapeutics: Many chemotherapy agents work by inducing apoptosis. BH3 mimetics (e.g., venetoclax) block BCL-2, restoring apoptosis in cancer cells.
Sources: Robbins & Kumar Basic Pathology, p. 24-26; Sleisenger and Fordtran's Gastrointestinal and Liver Disease, p. 23-24
This is a shared conversation. Sign in to Orris to start your own chat.