clinical features, Biochemical defect & Lab Diagnosis of Hartrupis disease (B) Albinism
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the complete answer.
A. Hartnup Disease
Biochemical Defect
Hartnup disease (OMIM #234500) is an autosomal recessive disorder caused by mutations in the SLC6A19 gene on chromosome 5p15, which encodes the neutral amino acid transporter B0AT1 (also called B°AT1). This transporter is located on the apical membrane of epithelial cells in the small intestine and renal proximal tubule.
The defect causes failure to absorb neutral (monoamino, monocarboxylic) amino acids from both the gut lumen and renal tubule. Importantly, the oligopeptide transporter is unaffected - so di- and tripeptides containing these amino acids are absorbed normally, which explains why only ~10% of patients develop overt symptoms.
Key pathway affected:
The critical consequence is tryptophan malabsorption. Tryptophan is normally converted to nicotinamide (niacin/Vitamin B3) via the kynurenine pathway (60 mg tryptophan → ~1 mg niacin). When tryptophan is lost, niacin cannot be synthesized endogenously, producing a functional niacin/pellagra-like deficiency state.
Note: The B0AT1 transporter requires either collectrin (in kidney) or ACE2 (in intestine - also the SARS-CoV-2 binding site) for surface expression.
Amino Acids Affected
The following neutral amino acids are malabsorbed and excreted in urine in quantities 5-10x normal:
| Group | Amino Acids |
|---|---|
| Neutral aliphatic | Alanine, Serine, Threonine, Valine, Leucine, Isoleucine |
| Neutral aromatic | Phenylalanine, Tyrosine, Tryptophan |
| Others | Glutamine, Asparagine, Histidine, Methionine |
NOT affected: Glycine, proline, hydroxyproline, cystine, dibasic amino acids (lysine, arginine, ornithine), and dicarboxylic amino acids (glutamate, aspartate).
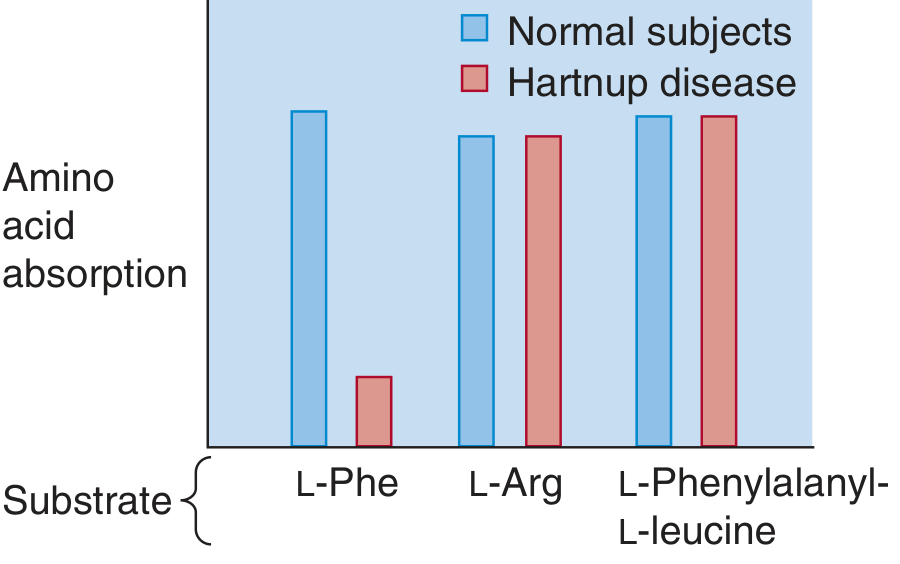
The bar chart below shows reduced intestinal absorption of neutral amino acids (e.g., L-Phe) in Hartnup disease, while cationic amino acids (L-Arg) and dipeptides (L-Phenylalanyl-L-leucine) are absorbed normally:
Clinical Features
The disease is named after the Hartnup family in England (first described 1956). It is the second most common inherited aminoaciduria after phenylketonuria, with an incidence of ~1 in 24,000-26,000.
Most patients identified by newborn screening are completely asymptomatic (oligopeptide absorption compensates). When symptoms occur, they are due to nicotinamide deficiency and present as:
1. Skin (Pellagra-like Dermatitis)
- Photosensitive erythematous, scaly rash on sun-exposed areas - face, neck, hands, legs
- Rash flares into hot, red, exudative state after sunlight exposure
- Followed by hyperpigmentation
- Stomatitis and vulvitis may occur
- Disease becomes milder with increasing age
- Rarely, an acrodermatitis enteropathica-like eruption may occur
2. Neurological
- Intermittent cerebellar ataxia (most characteristic neurologic sign)
- Spastic paraplegia (attacks)
- Emotional lability, psychosis, depression
- Frank delirium
- Rarely: mental retardation
3. Precipitating Factors
- Fever, sunlight, physical/emotional stress
- Sulfonamide therapy
- Inadequate dietary intake or increased metabolic demands (e.g., prolonged lactation)
"Hartnup disorder should be suspected in all subjects with pellagra and unexplained intermittent ataxia." - Brenner and Rector's The Kidney
Lab Diagnosis
1. Urinary Aminogram (Amino Acid Chromatography) - GOLD STANDARD
- Shows markedly increased excretion of neutral amino acids in the urine (5-10x normal)
- The pattern of excretion is the determining factor, not just the total amount
- The same 12-13 neutral amino acids are elevated in virtually all patients
- Normal levels of: glycine, proline, hydroxyproline, cystine, dibasic amino acids (Lys, Arg, Orn), and dicarboxylic amino acids
This distinguishes Hartnup disease from:
- Renal Fanconi syndrome (generalized aminoaciduria - all amino acids elevated)
- Dietary niacin deficiency/pellagra (no aminoaciduria)
2. Genetic Testing
- Sequencing of the SLC6A19 gene (or CTLRN gene coding collectrin, whose deficiency produces a biochemical phenocopy)
3. Urine Indole Compounds
- Excess unabsorbed tryptophan in the gut is metabolized by intestinal bacteria to indole derivatives (indican, indolylacetic acid), which are excreted in urine - detectable by urine indole tests
Summary Table
| Test | Finding in Hartnup |
|---|---|
| Urine amino acid chromatography | Elevated neutral amino acids (x5-10 normal) |
| Dibasic/dicarboxylic amino acids | Normal |
| Cystine in urine | Normal |
| Glycine in urine | Normal |
| Urine indoles | Elevated |
| Genetic test | SLC6A19 mutation |
Treatment: Nicotinamide (niacinamide) 50-300 mg/day + high-protein diet. Skin lesions respond well; neurologic disease may not always improve.
B. Albinism
Biochemical Defect
Albinism refers to a group of conditions in which a defect in tyrosine metabolism results in deficient or absent melanin production. The key enzyme is tyrosinase, a copper-requiring enzyme.
Normal Melanin Synthesis Pathway:
Tyrosine → (Tyrosinase) → DOPA → (Tyrosinase) → DOPAquinone → → → Melanin
Tyrosinase catalyzes two sequential reactions:
- Hydroxylation of tyrosine → L-DOPA (L-3,4-dihydroxyphenylalanine)
- Oxidation of L-DOPA → DOPAquinone
Both steps are blocked when tyrosinase is absent or defective, completely halting melanin biosynthesis.
Types of Albinism
Oculocutaneous Albinism (OCA) - Most Common Forms
OCA is autosomal recessive with absence/reduction of melanin in skin, hair, and eyes.
| Type | Gene | Defect | Features |
|---|---|---|---|
| OCA1A | TYR (tyrosinase) | Complete absence of tyrosinase activity | Most severe - white hair/skin, pink/red irises, VA 20/400 |
| OCA1B | TYR | Greatly reduced (not absent) tyrosinase | Some pigment develops with age, can tan |
| OCA2 | OCA2/P-gene | Defect in melanosomal membrane protein (P-protein); affects tyrosinase processing | Most common worldwide (~50% of OCA); pigmented hair at birth |
| OCA3 | TYRP1 | Tyrosinase-related protein 1 deficiency | "Rufous/red albinism" |
| OCA4 | SLC45A2 | Melanosomal transport protein | Similar to OCA2 |
- OCA1 (tyrosinase mutation): ~40% of OCA worldwide; most common in Japanese and Europeans
- OCA2 (P-gene mutation): ~50% of OCA worldwide; most common in sub-Saharan Africa (1:4,000)
Ocular Albinism (OA)
- X-linked inheritance (OA1: GPR143 gene mutation)
- Pigment defect confined primarily to the eyes (skin appears relatively normal)
Clinical Features
Cutaneous
- Absent or markedly reduced pigment in skin, hair, and eyebrows/lashes
- White to cream-colored hair (OCA1A: snow white hair; other types may have yellow to light brown hair)
- Skin does not tan (OCA1A); may develop minimal tan with age in OCA1B/OCA2
- Increased risk of skin cancer (basal cell, squamous cell carcinoma, melanoma) - major mortality risk
- Amelanotic nevi may be present
Ocular (All OCA types)
- Photophobia (sunlight hurts eyes) - due to absence of iris pigment allowing excess light
- Nystagmus (horizontal, pendular)
- Strabismus
- Reduced visual acuity (moderate to severe - 20/200 to 20/400 in OCA1A)
- Iris translucency - irises appear light blue to pink/red (OCA1A) due to visible blood vessels
- Foveal hypoplasia - lack of melanin during development leads to failure of proper foveal development
- Misrouting of optic nerve fibers (abnormal visual pathway development)
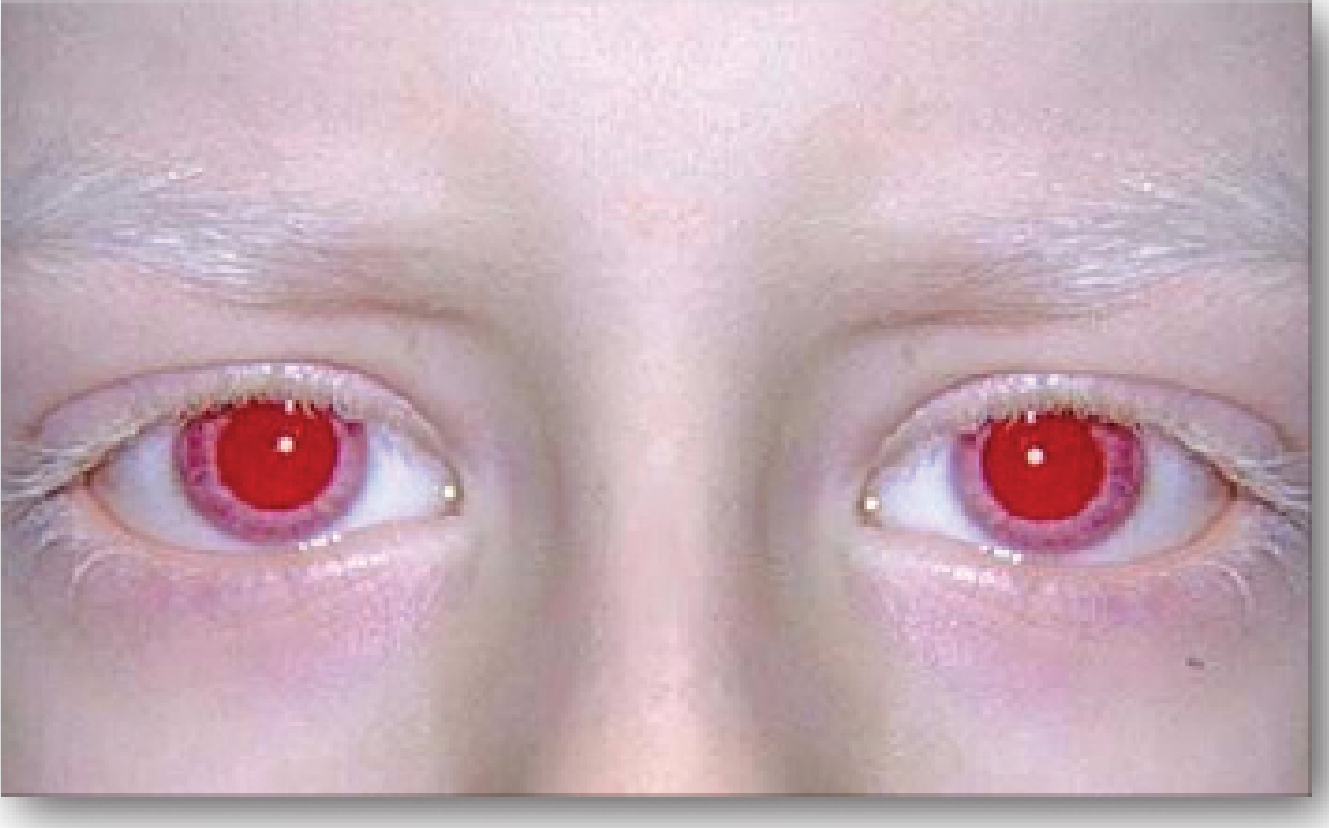
Below is a clinical photograph showing the characteristic red/pink irises and white eyebrows in oculocutaneous albinism:
Associated Syndromes
- Hermansky-Pudlak syndrome: OCA + platelet storage pool deficiency (bleeding) + ceroid accumulation
- Chediak-Higashi syndrome: OCA + recurrent infections + giant granules in leukocytes + peripheral neuropathy
- Griscelli syndrome: OCA + immunodeficiency + hemophagocytic lymphohistiocytosis
- Prader-Willi/Angelman syndromes: Associated with OCA2 (chromosome 15q11-13 locus)
Lab Diagnosis
| Test | Finding |
|---|---|
| Hair bulb tyrosinase test | OCA1A: tyrosinase-negative (hair bulbs do NOT darken in DOPA solution); OCA1B+: tyrosinase-positive (hair bulbs darken) |
| Genetic/molecular testing | Mutation analysis of TYR, OCA2, TYRP1, SLC45A2 genes - definitive diagnosis |
| Ophthalmic evaluation | Iris transillumination, nystagmus, visual evoked potentials (VEP show decussation abnormality) |
| Skin biopsy | Melanocytes present (not absent) but contain no melanin/premelanosomes only |
| Electron microscopy | Shows stage I/II premelanosomes; no mature melanosomes |
| Visual evoked potentials | Abnormal optic chiasm decussation pattern (useful in diagnosing OA) |
Key distinction: In albinism, melanocytes are present but non-functional (unlike vitiligo, where melanocytes are absent/destroyed).
Comparison Summary
| Feature | Hartnup Disease | Albinism (OCA) |
|---|---|---|
| Inheritance | Autosomal recessive | Autosomal recessive (primarily) |
| Gene | SLC6A19 (B0AT1 transporter) | TYR (tyrosinase) or others |
| Biochemical defect | Defective neutral amino acid transporter | Absent/defective tyrosinase (or melanosomal proteins) |
| Key missing metabolite | Niacin (via tryptophan) | Melanin |
| Skin findings | Pellagra-like photosensitive rash | Hypopigmentation, skin cancer risk |
| Eye findings | None specific | Photophobia, nystagmus, reduced VA |
| Neuro findings | Cerebellar ataxia, psychosis | None (except in syndromes) |
| Lab test | Urine neutral aminoaciduria | Hair bulb DOPA test; gene sequencing |
| Treatment | Nicotinamide + high-protein diet | Sun protection; no cure |
Sources: Harrison's Principles of Internal Medicine 22E; Brenner and Rector's The Kidney; Andrews' Diseases of the Skin; Lippincott's Biochemistry 8th ed; Medical Physiology (Boron); Emery's Medical Genetics
This is a shared conversation. Sign in to Orris to start your own chat.