Acute inflammation - explain this for 10 mark essay
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have all the content needed for a thorough 10-mark essay. Let me compose the full answer.
Acute Inflammation
Definition and Overview
Acute inflammation is a rapid, stereotyped host response to injury, infection, or necrosis. It is the early, non-specific arm of the immune response, characterized by vascular and cellular events that together aim to deliver leukocytes and plasma proteins to the site of damage. It typically develops within minutes to hours and resolves in days if the injurious agent is eliminated.
The cardinal signs, described by Celsus and Virchow, are:
- Rubor (redness)
- Calor (heat)
- Tumor (swelling)
- Dolor (pain)
- Functio laesa (loss of function)
Acute inflammation has three major components: (1) dilation of small vessels increasing blood flow; (2) increased vascular permeability enabling plasma proteins and leukocytes to leave the circulation; (3) emigration and activation of leukocytes at the focus of injury.
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 84)
1. Causes
Acute inflammation is triggered by:
- Infections (bacteria, viruses, fungi, parasites)
- Tissue necrosis (infarction, trauma, burns, chemical injury)
- Foreign bodies
- Immune reactions (hypersensitivity)
2. Vascular Reactions
Changes in Vascular Flow and Caliber
Shortly after injury, a transient vasoconstriction occurs (seconds), followed rapidly by vasodilation of arterioles and postcapillary venules - mediated primarily by histamine acting on vascular smooth muscle. This increases blood flow and accounts for the redness and heat.
Vasodilation is quickly followed by increased permeability with outpouring of protein-rich fluid (exudate) into the interstitium. Loss of fluid combined with increased vessel diameter leads to stasis (slowed blood flow) and red cell concentration in small vessels - seen histologically as vascular congestion.
Exudate vs. Transudate: An exudate is protein-rich extravascular fluid implying increased vascular permeability. A transudate is protein-poor, arising from osmotic/hydrostatic imbalance without increased permeability.
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 84)
Increased Vascular Permeability (Vascular Leakage)
Permeability is increased by several mechanisms:
- Endothelial cell contraction - the most common mechanism; induced by histamine, bradykinin, leukotrienes, and substance P acting on postcapillary venules; occurs within minutes (immediate transient response)
- Endothelial injury - direct damage by burns, toxins, or microbes causing immediate sustained response
- Leukocyte-mediated injury - activated neutrophils and monocytes release proteases and ROS that damage the endothelium
- Increased transcytosis - VEGF and other factors increase transport across endothelium
- Leakage from new blood vessels during repair (angiogenesis)
3. Cellular Events - Leukocyte Recruitment
The journey of leukocytes from vessel lumen to tissue follows a defined multistep sequence:
Step 1: Margination and Rolling
As blood flow slows (stasis), leukocytes (primarily neutrophils) redistribute to the periphery - a process called margination. They then begin rolling along the endothelium, mediated by selectins:
- P-selectin: rapidly redistributed from Weibel-Palade bodies to the endothelial surface on stimulation by histamine/thrombin
- E-selectin: induced on endothelium by TNF and IL-1 within 1-2 hours
- L-selectin: on leukocytes, binds endothelial ligands
Step 2: Firm Adhesion
Rolling leukocytes are activated by chemokines displayed on endothelial surfaces, increasing the affinity of leukocyte integrins (LFA-1, MAC-1) for their endothelial ligands (ICAM-1, VCAM-1). This converts rolling to firm adhesion. TNF and IL-1 upregulate ICAM-1 and VCAM-1 on endothelium.
Step 3: Transmigration (Diapedesis)
Leukocytes migrate through the endothelial junctions (paracellular route), driven by PECAM-1 (CD31) interactions. They then cross the basement membrane assisted by collagenases and move into the interstitium.
Step 4: Chemotaxis
Leukocytes migrate along a concentration gradient of chemotactic agents:
- Exogenous: bacterial products (N-formyl methionine peptides)
- Endogenous: C5a, leukotriene B4 (LTB4), IL-8 (CXCL8), chemokines
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 88-90)
4. Leukocyte Activation and Phagocytosis
Once at the site, leukocytes are activated by microbial products and local mediators to perform their primary functions:
Phagocytosis
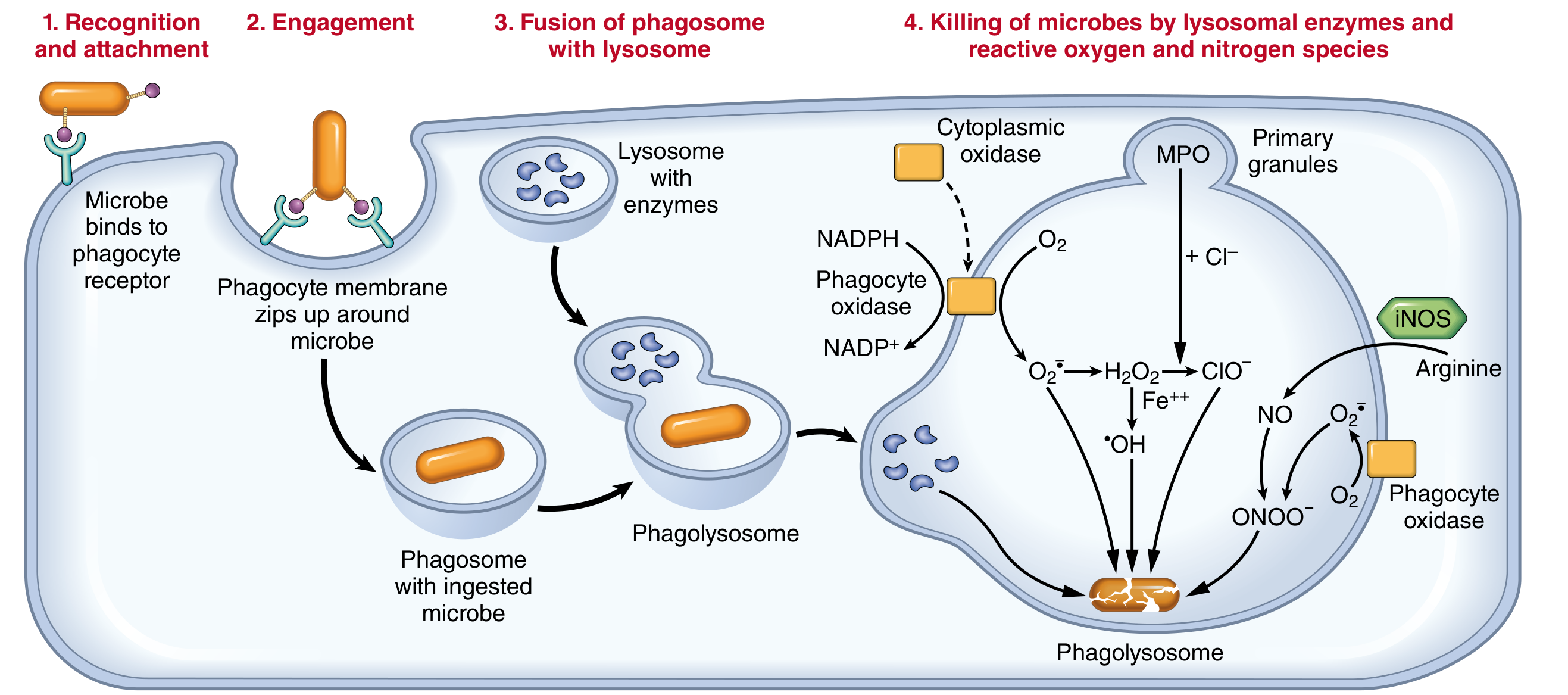
Phagocytosis proceeds in three steps:
- Recognition and attachment - enhanced by opsonins (IgG, C3b) binding to Fc receptors and complement receptors on leukocytes
- Engulfment - cytoplasmic extensions flow around the particle forming a phagosome, which fuses with lysosomes to form a phagolysosome
- Killing and degradation
Intracellular Killing
- Reactive oxygen species (ROS): NADPH oxidase assembles in the phagosome membrane, oxidizing NADPH and generating superoxide (O2-) - the respiratory burst. Superoxide is converted to H2O2 and then to hypochlorous acid (HOCl) by myeloperoxidase (MPO), the most potent bactericidal mechanism in neutrophils.
- Reactive nitrogen species: Inducible nitric oxide synthase (iNOS) generates NO, which reacts with superoxide to form peroxynitrite (ONOO-)
- Lysosomal enzymes: elastase, cathepsins, defensins, lysozyme
Neutrophil Extracellular Traps (NETs)
Activated neutrophils can extrude nuclear chromatin and antimicrobial proteins as extracellular nets that trap and kill microbes outside the cell.
5. Mediators of Inflammation
| Mediator | Source | Principal Action |
|---|---|---|
| Histamine | Mast cells, basophils | Vasodilation, increased permeability |
| Serotonin | Platelets | Vasodilation, increased permeability |
| Prostaglandins (PGE2, PGI2) | All cells (via COX) | Vasodilation, fever, pain |
| Leukotrienes (LTB4) | Leukocytes (via 5-LOX) | Chemotaxis |
| LTC4, LTD4, LTE4 | Mast cells, leukocytes | Increased permeability, bronchoconstriction |
| C3a, C5a | Complement cascade | Chemotaxis, opsonization, mast cell degranulation |
| TNF, IL-1 | Macrophages, mast cells | Endothelial activation, fever, acute-phase response |
| IL-8 (CXCL8) | Macrophages, endothelium | Neutrophil chemotaxis |
| Bradykinin | Kinin system (factor XII) | Vasodilation, pain, increased permeability |
| PAF | Leukocytes, endothelium | Platelet aggregation, leukocyte activation |
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, Table 3.8)
Note: Prostaglandins are the main mediators of fever (via hypothalamic COX-2) and pain - which is why NSAIDs (COX inhibitors) reduce both. Corticosteroids inhibit phospholipase A2, blocking arachidonic acid release and thus the synthesis of both prostaglandins and leukotrienes.
6. Morphologic Patterns of Acute Inflammation
The hallmarks on histology are dilated blood vessels and neutrophilic infiltration. Specific patterns depending on etiology and site:
| Pattern | Features | Example |
|---|---|---|
| Serous | Watery, protein-poor exudate; no significant neutrophils | Skin blister, pleural effusion in viral pleuritis |
| Fibrinous | Large protein exudate with fibrin deposition; "bread and butter" pericarditis | Acute pericarditis, lobar pneumonia |
| Purulent (Suppurative) | Abundant pus (neutrophils + dead cells + microbes); abscess formation | Bacterial pneumonia, brain abscess |
| Ulcer | Local defect of epithelial surface; necrosis of inflamed tissue | Peptic ulcer, pressure sore |
7. Outcomes of Acute Inflammation
Acute inflammation has three possible outcomes:
- Complete resolution - the usual outcome when injury is limited and parenchyma can regenerate; involves macrophage clearance of debris and lymphatic resorption of edema
- Healing by scarring (fibrosis/organization) - when tissue destruction is extensive, or the tissue cannot regenerate (e.g., cardiac muscle); fibrin exudate is organized by connective tissue ingrowth
- Progression to chronic inflammation - when the injurious agent persists or normal healing is impaired; characterized by shift from neutrophils to mononuclear infiltrate (macrophages, lymphocytes)
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 105)
Summary
Acute inflammation is a tightly coordinated defensive response involving:
- Vascular phase: vasodilation and increased permeability causing exudate formation
- Cellular phase: leukocyte margination, rolling (selectins), firm adhesion (integrins/ICAM), transmigration (PECAM-1), chemotaxis, and phagocytosis
- Mediator cascade: histamine, eicosanoids, cytokines, complement, and kinins amplify and direct the response
- Resolution or progression: complete healing if the agent is eliminated; scarring or chronic inflammation if not
The cardinal signs - rubor, calor, tumor, dolor, and functio laesa - arise directly from the vascular and cellular changes: increased blood flow causes redness and heat; exudate causes swelling; prostaglandins and bradykinin cause pain; and tissue damage plus pain cause loss of function.
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.), Chapter 3 - Inflammation and Repair
This is a shared conversation. Sign in to Orris to start your own chat.