
9C778AB9-79B8-4E60-B752-2AF02FF13003.png
Explain in details
Reading File
analyse_media
Finding Sources
Finding Sources
Reading File
Reading File
I now have rich content from the diagram analysis and the textbooks. Let me compose the full detailed explanation.
Acute Coronary Syndrome (ACS): Full Detailed Explanation
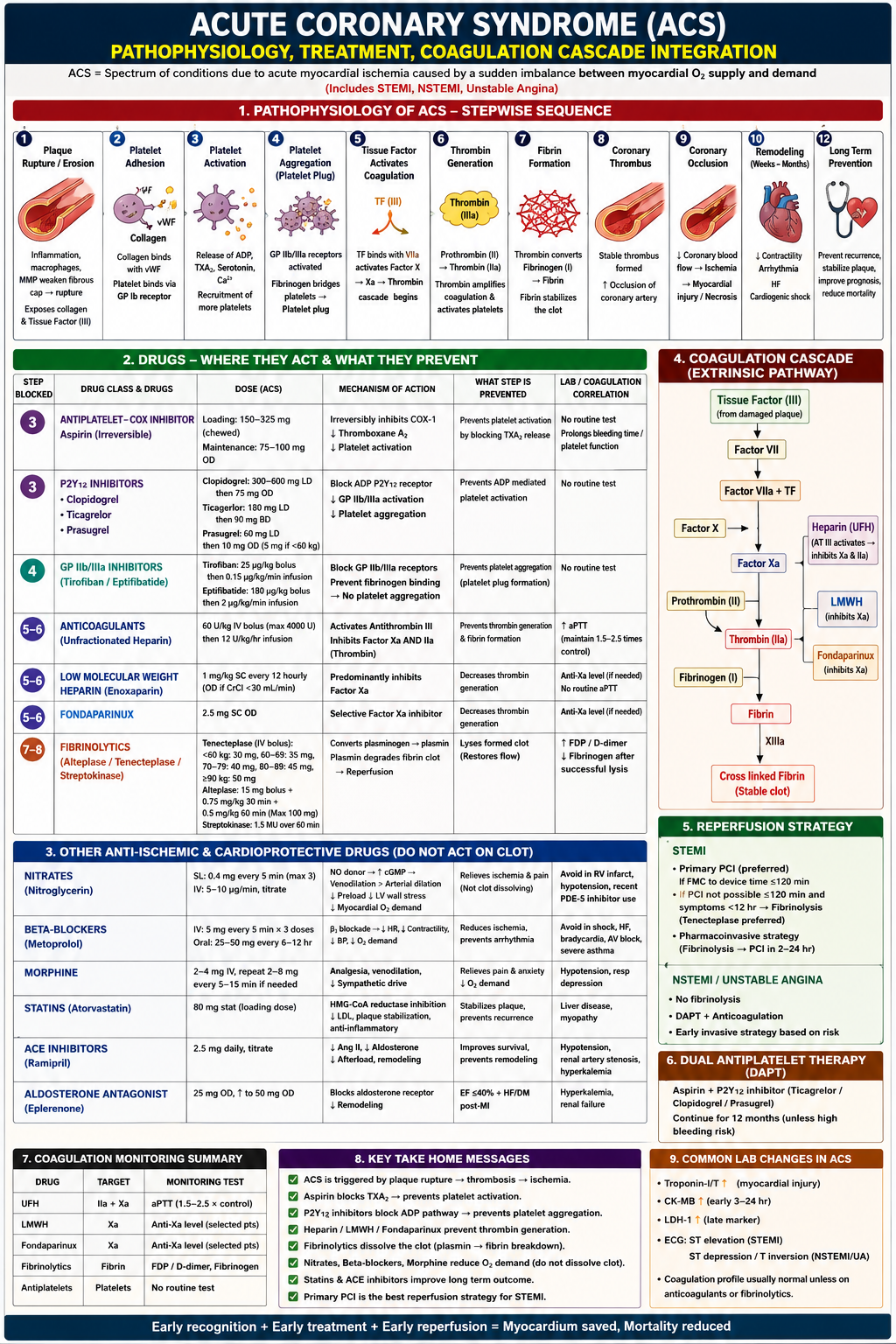
What is ACS?
ACS is a spectrum of ischemic myocardial conditions caused by a sudden imbalance between myocardial oxygen supply and demand. It includes three clinical entities:
- STEMI - ST-Elevation Myocardial Infarction (complete occlusion)
- NSTEMI - Non-ST-Elevation Myocardial Infarction (subtotal occlusion)
- Unstable Angina - ischemia without myocyte necrosis (troponin negative)
As Goldman-Cecil Medicine explains, the key distinction is that NSTEMI and UA both lack persistent ST elevation and are collectively termed "non-ST elevation ACS," while STEMI involves persistent (>20 min) ST elevation with confirmed myonecrosis.
Section 1: Pathophysiology - The 12-Step Sequence
This is the core of the diagram. Every drug targets a specific step here.
Step 1 - Plaque Rupture / Erosion
- Atherosclerotic plaques that are vulnerable have a large lipid core, thin fibrous cap (≤65 µm), high macrophage density, and matrix metalloproteinase (MMP) activity
- MMPs secreted by macrophages and foam cells degrade the fibrous cap, causing it to rupture or erode
- This exposes two critical pro-thrombotic substances beneath: Collagen and Tissue Factor (TF, Factor III)
- This is the triggering event for the entire cascade
Step 2 - Platelet Adhesion
- Exposed subendothelial collagen binds von Willebrand Factor (vWF), which acts as a molecular bridge
- Circulating platelets recognize this collagen-vWF complex and bind to it via their GP Ib receptors (Glycoprotein Ib)
- This is the initial, reversible attachment of platelets to the damaged vessel wall
Step 3 - Platelet Activation
- Adhered platelets become activated and release a burst of chemical signals:
- ADP (Adenosine Diphosphate) - recruits more platelets
- Thromboxane A2 (TXA2) - potent vasoconstrictor and platelet recruiter (synthesized via COX-1)
- Serotonin - promotes vasoconstriction
- Ca2+ - intracellular signaling mediator
- These signals cause conformational changes in the platelet and recruit more platelets in a positive feedback loop
This is where Aspirin and P2Y12 inhibitors act - by blocking TXA2 production and ADP signaling respectively.
Step 4 - Platelet Aggregation (Platelet Plug)
- Activated platelets expose GP IIb/IIIa receptors on their surface
- Fibrinogen molecules bridge adjacent platelets by binding GP IIb/IIIa receptors simultaneously on two platelets
- This forms the primary hemostatic plug (soft, unstable platelet plug)
This is where GP IIb/IIIa inhibitors act - blocking the final common pathway of aggregation.
Step 5 - Tissue Factor Activates Coagulation (Extrinsic Pathway)
- Exposed Tissue Factor (TF/Factor III) from the ruptured plaque binds with circulating Factor VIIa to form the TF-VIIa complex
- This complex activates Factor X → Factor Xa
- Factor Xa combines with Factor Va (prothrombinase complex) to convert Prothrombin to Thrombin
- This is the start of the coagulation cascade (extrinsic pathway)
Step 6 - Thrombin Generation
- Prothrombin (Factor II) → Thrombin (Factor IIa) via the prothrombinase complex
- Thrombin is the central enzyme of coagulation - it does three things simultaneously:
- Converts fibrinogen to fibrin
- Amplifies coagulation by activating more upstream factors (V, VIII, XI)
- Directly activates platelets via PAR-1 receptors (further fueling aggregation)
Heparins and Fondaparinux act at steps 5-6 by inhibiting Factor Xa and/or Thrombin (IIa).
Step 7 - Fibrin Formation
- Thrombin cleaves soluble Fibrinogen (Factor I) into insoluble Fibrin monomers
- Factor XIIIa (activated by Thrombin) cross-links these fibrin monomers into a stable fibrin mesh
- This reinforces the platelet plug, forming a stable, firm thrombus
Step 8 - Coronary Thrombus
- The combined platelet plug + cross-linked fibrin = coronary thrombus
- In STEMI: complete occlusion
- In NSTEMI/UA: subtotal (partial) occlusion
Step 9 - Coronary Occlusion → Ischemia → Necrosis
- Blocked coronary blood flow → ischemia → if prolonged, irreversible myocyte necrosis (infarction)
- Troponin I and T are released from dying myocytes into the bloodstream
- CK-MB rises within 3-24 hours; LDH-1 is a late marker
Step 10 - Remodeling (Weeks to Months)
- Infarcted myocardium is replaced by scar tissue
- Adverse remodeling: decreased contractility, ventricular dilatation, arrhythmias, heart failure, cardiogenic shock
- ACE inhibitors, Beta-blockers, and Aldosterone antagonists are given to prevent/slow remodeling
Step 12 - Long-Term Prevention
- Statins stabilize remaining plaques and reduce LDL
- DAPT (Dual Antiplatelet Therapy), ACE inhibitors, and beta-blockers continue for secondary prevention
Section 2: Drugs - Where They Act
A. Antiplatelet Drugs
1. Aspirin (COX Inhibitor) - Acts at Step 3
| Feature | Detail |
|---|---|
| Loading Dose | 150-325 mg chewed (for rapid absorption) |
| Maintenance | 75-100 mg OD |
| Mechanism | Irreversibly inhibits COX-1 enzyme → blocks TXA2 synthesis → less platelet activation |
| Duration | Irreversible; platelet lifespan is 7-10 days, so effect lasts until new platelets are formed |
| Lab Test | No routine test; prolongs bleeding time |
Key point: aspirin is "irreversible" - this is why it must be stopped 7-10 days before surgery, not just 24 hours.
2. P2Y12 Inhibitors - Acts at Step 3
These block the ADP receptor on platelets, preventing ADP-mediated activation and downstream GP IIb/IIIa activation.
| Drug | Loading Dose | Maintenance | Notes |
|---|---|---|---|
| Clopidogrel | 300-600 mg LD | 75 mg OD | Prodrug, requires CYP2C19 activation; variable response |
| Ticagrelor | 180 mg LD | 90 mg BD | Direct-acting (not prodrug); faster onset; preferred in STEMI |
| Prasugrel | 60 mg LD | 10 mg OD (5 mg if <60 kg) | Prodrug but more reliably activated; avoid in prior stroke/TIA |
3. GP IIb/IIIa Inhibitors - Acts at Step 4
Blocks the final common pathway of platelet aggregation:
| Drug | Dose | Note |
|---|---|---|
| Tirofiban | 25 µg/kg bolus, then 0.15 µg/kg/min | Small molecule; reversible |
| Eptifibatide | 180 µg/kg bolus, then 2 µg/kg/min | Cyclic peptide |
These are used mainly in high-risk NSTEMI/UA undergoing PCI.
B. Anticoagulants - Act at Steps 5-6
4. Unfractionated Heparin (UFH)
- Dose: 60 U/kg IV bolus (max 4000 U), then 12 U/kg/hr infusion
- Mechanism: Activates Antithrombin III, which then inhibits both Factor Xa AND Factor IIa (Thrombin)
- Monitoring: aPTT maintained at 1.5-2.5 times control
- Advantage: Reversible with protamine sulfate; adjustable by infusion
5. Low Molecular Weight Heparin (LMWH) - Enoxaparin
- Dose: 1 mg/kg SC every 12 hours (once daily if CrCl <30 mL/min)
- Mechanism: Predominantly inhibits Factor Xa (less Thrombin inhibition than UFH)
- Monitoring: Anti-Xa level only when needed (e.g., renal impairment, obesity, pregnancy); no routine aPTT needed
- Advantage: Predictable pharmacokinetics, subcutaneous dosing, less HIT risk
6. Fondaparinux
- Dose: 2.5 mg SC OD
- Mechanism: Selective, direct Factor Xa inhibitor (via Antithrombin III)
- Advantage: No HIT (Heparin-Induced Thrombocytopenia) risk; preferred in NSTEMI/UA where PCI is not immediately planned
C. Fibrinolytics (Thrombolytics) - Act at Steps 7-8
These dissolve already formed clots by converting plasminogen to plasmin, which breaks down the fibrin mesh.
| Drug | Dose | Notes |
|---|---|---|
| Tenecteplase (TNK) | Weight-based IV bolus: <60 kg=30 mg, 60-69 kg=35 mg, 70-79 kg=40 mg, 80-89 kg=45 mg, ≥90 kg=50 mg | Single bolus; most fibrin-specific; preferred |
| Alteplase (tPA) | 15 mg bolus + 0.75 mg/kg over 30 min + 0.5 mg/kg over 60 min (max 100 mg) | Infusion; fibrin-specific |
| Streptokinase | 1.5 MU over 60 min | Non-fibrin-specific; risk of allergic reactions; cheapest |
Lab changes after successful fibrinolysis:
- FDP/D-dimer rises (fibrin breakdown products released)
- Fibrinogen falls (consumed by plasmin)
ONLY used in STEMI when PCI is not available within 120 minutes of first medical contact.
Section 3: Other Anti-Ischemic & Cardioprotective Drugs (Do NOT dissolve clot)
These drugs reduce myocardial oxygen demand or promote long-term healing - they don't act on the thrombus.
Nitrates (Nitroglycerin)
- SL: 0.4 mg every 5 min, max 3 doses
- IV: 5-10 µg/min, titrate up
- MOA: Nitric oxide (NO) donor → increases cGMP → venodilation > arterial dilation → reduces preload → decreases LV wall stress → reduces myocardial O2 demand
- Also: Dilates coronary arteries (relieves spasm)
- Avoid: RV infarction (preload-dependent), hypotension (SBP <90), recent PDE-5 inhibitor use (sildenafil/tadalafil - can cause fatal hypotension)
Beta-Blockers (Metoprolol)
- IV: 5 mg every 5 min × 3 doses; Oral: 25-50 mg every 6-12 hours
- MOA: Beta-1 blockade → decreased HR, decreased contractility, decreased BP → reduced O2 demand + anti-arrhythmic effect
- Benefit: Reduces infarct size, prevents ventricular arrhythmias, reduces mortality
- Avoid: Acute decompensated HF, cardiogenic shock, bradycardia (<60 bpm), AV block (>1st degree), severe asthma/COPD
Morphine
- Dose: 2-4 mg IV, repeat 2-8 mg every 5-15 min if needed
- MOA: Analgesia + venodilation (reduces preload) + reduces sympathetic drive (lowers HR and BP)
- Effect: Relieves pain and anxiety, reduces O2 demand
- Note: Recent data suggests morphine may delay absorption of oral P2Y12 inhibitors (due to slowed gastric emptying) - use cautiously
Statins (Atorvastatin)
- Dose: 80 mg stat (high-intensity loading dose)
- MOA: HMG-CoA reductase inhibition → decreased LDL + plaque stabilization + anti-inflammatory effects
- Effect: Prevents plaque recurrence, stabilizes vulnerable plaques
- Lab: Monitor liver disease, myopathy (CK if symptomatic)
ACE Inhibitors (Ramipril)
- Dose: 2.5 mg daily, titrate up
- MOA: Blocks angiotensin II and aldosterone → decreased afterload → prevents adverse cardiac remodeling
- Benefit: Improves survival, especially in anterior MI, EF <40%, DM, hypertension
- Avoid: Hypotension, renal artery stenosis, hyperkalemia
Aldosterone Antagonist (Eplerenone)
- Dose: 25 mg OD, titrate to 50 mg OD
- Indication: EF ≤40% + Heart Failure OR post-MI
- MOA: Blocks aldosterone receptor → reduces myocardial fibrosis and remodeling
- Avoid: Hyperkalemia, renal failure
Section 4: Coagulation Cascade (Extrinsic Pathway) and Drug Targets
The right-side diagram shows the extrinsic coagulation pathway and exactly where each anticoagulant acts:
Tissue Factor (TF, from damaged plaque)
↓
Factor VII
↓
Factor VIIa + TF complex
↓
Factor X → Factor Xa ←── LMWH inhibits here
↓ ←── Fondaparinux inhibits here
Prothrombin (II) ←── Heparin (UFH) inhibits BOTH Xa AND IIa
↓
Thrombin (IIa) ←── UFH also inhibits here
↓
Fibrinogen (I)
↓
Fibrin
↓
Cross-linked Fibrin (stable clot) ← via Factor XIIIa
Key distinction:
- UFH: Inhibits both Xa and IIa (via Antithrombin III)
- LMWH: Predominantly inhibits Xa
- Fondaparinux: Selectively inhibits Xa ONLY
Section 5: Reperfusion Strategy
STEMI
- Primary PCI (Percutaneous Coronary Intervention) is always preferred - balloon/stent opens the blocked artery mechanically
- Target: FMC (First Medical Contact) to device time ≤120 minutes
- If PCI not achievable within 120 min AND symptoms <12 hours → Fibrinolysis (Tenecteplase)
- After fibrinolysis → Pharmacoinvasive strategy: transfer to PCI center for angiography within 2-24 hours
NSTEMI / Unstable Angina
- No fibrinolytics (the clot is subtotal; lysis carries bleeding risk without proven benefit)
- DAPT + Anticoagulation (medical stabilization)
- Early invasive strategy based on risk stratification (GRACE score, TIMI score)
Section 6: Dual Antiplatelet Therapy (DAPT)
Aspirin + a P2Y12 inhibitor (Ticagrelor or Clopidogrel or Prasugrel)
- Ticagrelor is preferred in most ACS (faster, more potent, better outcomes in PLATO trial)
- Duration: Continue for 12 months post-ACS (unless high bleeding risk)
- After 12 months, aspirin monotherapy continues indefinitely
Section 7: Coagulation Monitoring Summary
| Drug | Target | Monitoring Test |
|---|---|---|
| UFH | Factors IIa + Xa | aPTT (maintain 1.5-2.5 × control) |
| LMWH | Factor Xa | Anti-Xa level (selected patients) |
| Fondaparinux | Factor Xa | Anti-Xa level (selected patients) |
| Fibrinolytics | Fibrin | FDP/D-dimer, Fibrinogen |
| Antiplatelets | Platelets | No routine test |
Section 9: Common Lab Changes in ACS
| Test | Change | Timing/Notes |
|---|---|---|
| Troponin I/T | Rises | First to rise; most sensitive/specific for myocardial injury |
| CK-MB | Rises | Early 3-24 hours; used to detect re-infarction |
| LDH-1 | Rises late | Late marker; peaks at 3-5 days |
| ECG | ST elevation (STEMI) or ST depression/T-wave inversion (NSTEMI/UA) | |
| Coagulation profile | Usually normal | Abnormal only if on anticoagulants or fibrinolytics |
Key Takeaway Messages (Bottom of Diagram)
- ACS is triggered by plaque rupture → thrombosis → ischemia
- Aspirin blocks TXA2 → prevents platelet activation
- P2Y12 inhibitors block ADP pathway → prevents platelet aggregation
- Heparin/LMWH/Fondaparinux prevent thrombin generation
- Fibrinolytics dissolve the clot (plasmin → fibrin breakdown) - only in STEMI when PCI unavailable
- Nitrates, Beta-blockers, Morphine reduce O2 demand - do NOT dissolve clot
- Statins and ACE inhibitors improve long-term outcomes
- Primary PCI is the best reperfusion strategy for STEMI
The overarching principle: Each drug class targets a different step in the pathophysiology cascade - from plaque rupture all the way to long-term remodeling. Combining them (e.g., DAPT + anticoagulation + statin + beta-blocker + ACE inhibitor) provides maximal protection at each stage. As stated in Goldman-Cecil Medicine and Fuster and Hurst's The Heart, 15th Ed: early recognition + early treatment + early reperfusion = myocardium saved, mortality reduced.
This is a shared conversation. Sign in to Orris to start your own chat.