SSPE etiology, clinical feature , stages , radiological feature , treatment for standard sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Searching PubMed
"subacute sclerosing panencephalitis" treatment
I now have comprehensive information from multiple standard sources (Bradley & Daroff's Neurology, Adams & Victor's, Harrison's, Robbins Pathology, Goldman-Cecil Medicine). Let me now compile the full structured answer.
Subacute Sclerosing Panencephalitis (SSPE)
Etiology
SSPE is caused by persistent, non-productive (non-permissive) infection of neurons and glial cells by measles virus (a paramyxovirus). The key pathogenic mechanisms are:
- Defective virus maturation: The measles virus undergoes mutational changes that affect its M (matrix) protein and other envelope proteins, preventing proper virion assembly and budding. The virus cannot escape the cell and spreads only by direct cell-to-cell contact.
- Intracellular concealment: Because no free virus is released, the virus is largely hidden from the immune system despite very high serum and CSF antimeasles antibody titers (notably, antibody to the M protein is frequently absent).
- Epidemiology: SSPE is a rare late complication with a frequency estimated at 1 in 100,000-500,000 measles cases. The annual incidence is <0.1 to 5-6 cases per million in unimmunized populations. It has declined dramatically since measles vaccination was introduced but may rise with increasing vaccine hesitancy.
- Risk factors: Primary measles infection before 2 years of age (highest risk), male sex (M:F ratio ~3:1), immunocompetent host (unlike measles inclusion body encephalitis, which affects immunocompromised patients).
- Latency interval: Median 6-8 years after primary measles infection (range 2-12 years), followed by insidious neurological onset.
(Bradley & Daroff's Neurology in Clinical Practice; Harrison's Principles of Internal Medicine 22E; Robbins Pathologic Basis of Disease)
Clinical Features
SSPE presents as a progressive neurodegenerative disorder without the typical signs of CNS infection (no fever, no meningismus). 85% of patients are between 5 and 15 years old at diagnosis.
General Sequence
| Feature | Detail |
|---|---|
| Prior measles | Usually at age <2 years |
| Latent interval | 6-8 years (range 2-12 years) |
| Age at onset | 5-15 years (most common) |
| Sex | Males predominate (3:1) |
Stage-wise Clinical Progression (Jabbour Classification - 4 Stages)
Stage I - Insidious/Behavioral (weeks to months)
- Decline in school performance and academic ability
- Personality and mood changes, temper outbursts
- Difficulty with language, forgetfulness
- Loss of interest in usual activities
- No obvious neurological signs
Stage II - Neurological (weeks to months)
- Severe progressive intellectual deterioration
- Myoclonus - repetitive, shock-like jerks (characteristic and often leads to diagnosis)
- Focal and/or generalized seizures
- Ataxia, choreoathetoid or ballistic movements
- Chorioretinitis, visual disturbances
- Spasticity, hyperactive deep tendon reflexes
Stage III - Advanced/Coma
- Optic atrophy
- Quadriparesis, progressive unresponsiveness
- Autonomic instability
- Akinetic mutism
- Coma ("decerebrate/decorticate" posture)
Stage IV - Terminal/Vegetative
- Child lies insensate, virtually "decorticated"
- Complete loss of cortical function
- Death typically within 1-3 years of onset
Clinical course is usually steadily progressive to death within 1-3 years. Spontaneous remissions are estimated in ~5% of cases. Acute symptoms with increased intracranial pressure are poor prognostic signs.
(Bradley & Daroff's Neurology; Adams & Victor's Principles of Neurology 12E)
Histopathology (Pathological Features)
- Cowdry type A eosinophilic intranuclear inclusions in neurons and glial cells - the histopathological hallmark
- Cytoplasmic inclusions also present in neurons (cigar-shaped)
- Destruction of nerve cells, neuronophagia
- Perivenous cuffing by lymphocytes and mononuclear cells
- White matter: degeneration of myelinated fibers (both myelin and axons) + fibrous gliosis ("sclerosing")
- Distribution: cerebral cortex and white matter of both hemispheres + brainstem; cerebellum usually spared
- Electron microscopy shows measles nucleocapsids within inclusion-bearing cells
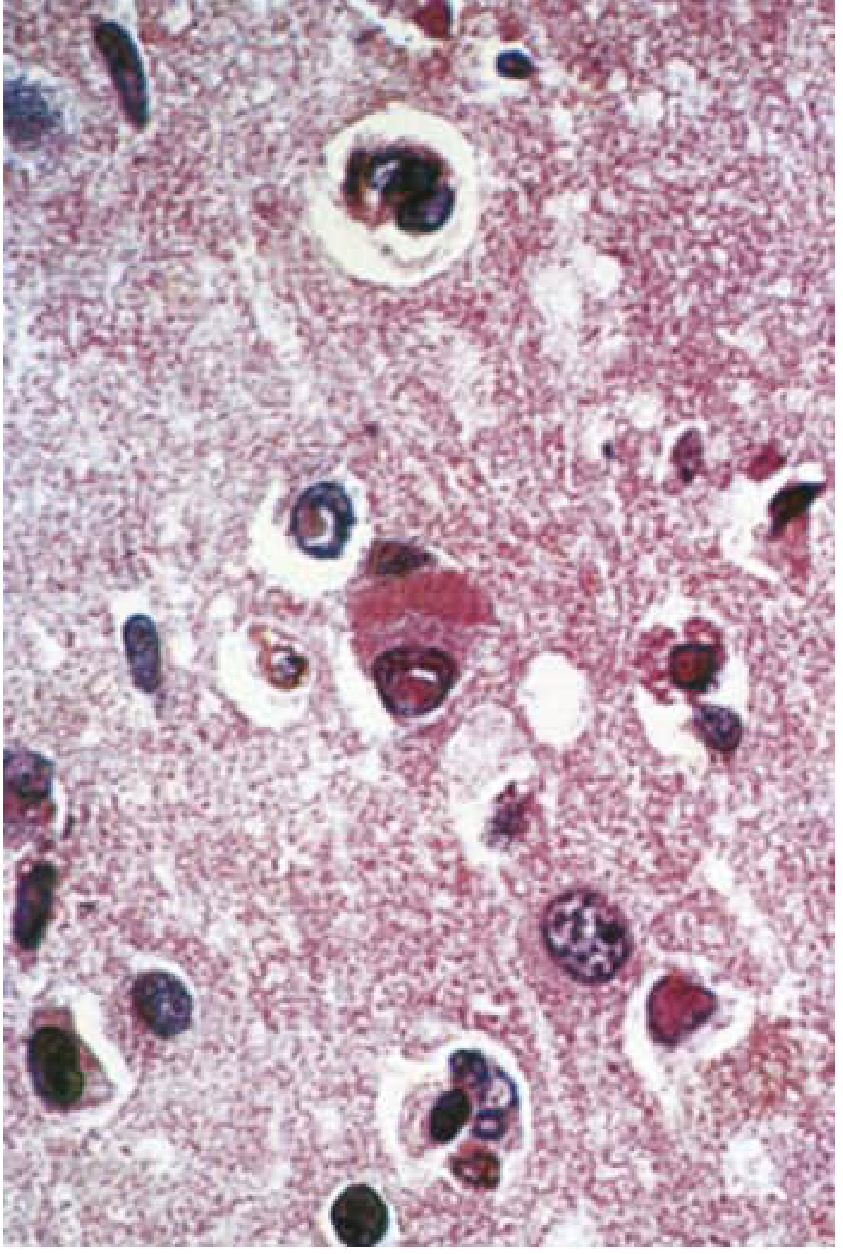
(Robbins Pathologic Basis of Disease; Bradley & Daroff's Neurology; Adams & Victor's)
Radiological Features (MRI/CT)
MRI (Investigation of Choice)
| Stage | MRI Findings |
|---|---|
| Early | Often normal; earliest changes are high T2/FLAIR signal in gray matter and subcortical white matter of posterior hemispheres (parieto-occipital region) |
| Stage II | T2/FLAIR hyperintensities extend - white matter involvement spreads anteriorly; cortical and subcortical lesions in temporal, parietal lobes |
| Advanced | Periventricular involvement; brainstem lesions; progressive cerebral atrophy (cortical and subcortical); white matter changes become confluent |
| Terminal | Widespread cortical atrophy; severe diffuse white matter changes |
- Lesions are asymmetric early, becoming more diffuse with progression
- No contrast enhancement (blood-brain barrier usually intact)
- Brainstem changes appear in later stages
- MRI changes begin in subcortical white matter and spread to the periventricular region
(Harrison's 22E; Bradley & Daroff's Neurology; Adams & Victor's)
EEG (Critical Diagnostic Tool)
- Pathognomonic pattern: Periodic (every 3-14 seconds, typically 5-8 seconds) bursts of high-voltage, sharp slow-wave complexes (2-3/s) lasting up to 3 seconds, against a background of depressed ("flat") activity
- The periodic complexes are synchronous with the myoclonic jerks
- Early SSPE may show only nonspecific slowing
- These periodic complexes appear prominently in Stage II
CSF Findings
- Acellular (no pleocytosis)
- Normal or mildly elevated protein
- Markedly elevated gamma globulin (>20% of total CSF protein)
- Oligoclonal bands (measles-virus-specific IgG)
- Elevated CSF antimeasles antibody - invariably elevated; CSF/serum ratio confirms intrathecal synthesis
- Antibody to M protein frequently absent
Diagnosis
- Clinical: Characteristic clinical stages in a child with history of early measles
- EEG: Periodic high-voltage slow-wave complexes
- CSF: Elevated measles antibody, oligoclonal bands, elevated gamma globulin
- Serum: Very high antimeasles antibody titers
- MRI: T2/FLAIR white matter changes (posterior > anterior early)
- Brain biopsy (if needed): Viral inclusions + measles antigen by immunocytochemistry, viral genome by PCR/in situ hybridization; measles virus can be cultured from brain by special cocultivation techniques
Treatment
No definitive, curative therapy exists. Management is largely supportive with some agents showing partial benefit.
Antiviral/Immunomodulatory Therapy
| Drug | Dose/Route | Remarks |
|---|---|---|
| Isoprinosine (Inosiplex) | 100 mg/kg/day orally (max 3 g/day) in 3 divided doses | Alone or combined with IFN-α; reported to prolong survival and produce clinical improvement in some patients; never subjected to controlled clinical trial |
| Intraventricular Interferon-α (via Ommaya reservoir) | 100,000 U/m² BSA/day, escalating to 10⁶ U/m²/day over 5 days; then 10⁶ U/m² twice weekly for 6 months | Combined with oral isoprinosine; ~30-35% of patients improved or stabilized |
| Subcutaneous Interferon-α | Up to 5 million units/day | Used to treat peripheral reservoirs (lymphoid, glandular tissue) simultaneously with intrathecal IFN |
| Ribavirin (IV) | In combination with intrathecal IFN-α | Limited case reports; some benefit reported |
| Amantadine | Oral | Used historically; improvement not consistently corroborated |
Symptomatic Treatment
- Anticonvulsants: e.g., Levetiracetam (shown to improve myoclonus and encephalopathy symptomatically)
- Seizure control, management of spasticity, nutritional support
Treatment Monitoring
- Laboratory endpoint: eradication of detectable measles antigen from CSF
- Risks of prolonged/repeated interferon: meningitis, IFN-α-induced encephalopathy, upper and lower motor neuron toxicity
Prevention (Most Effective Intervention)
- MMR vaccine (measles, mumps, rubella) at 12-15 months, second dose at 4-6 years (MMRV is the quadrivalent option)
- Vaccination essentially eliminates the risk of SSPE; the disease has practically disappeared in countries with high measles vaccination coverage
(Harrison's Principles of Internal Medicine 22E; Bradley & Daroff's Neurology in Clinical Practice; Adams & Victor's Principles of Neurology 12E)
Summary Table
| Parameter | Key Point |
|---|---|
| Cause | Persistent defective measles virus infection of CNS |
| Latency | 6-8 years after primary measles |
| Peak age | 5-15 years, boys > girls (3:1) |
| Stage I | Behavioral, school decline |
| Stage II | Myoclonus, seizures, ataxia, chorioretinitis |
| Stage III | Quadriparesis, akinetic mutism, coma |
| Stage IV | Vegetative/decorticate, death |
| EEG | Periodic high-voltage slow waves every 3-14 sec |
| MRI | T2/FLAIR: posterior subcortical > white matter > diffuse atrophy |
| CSF | Acellular, elevated IgG, antimeasles antibody, oligoclonal bands |
| Hallmark histology | Cowdry type A intranuclear inclusions |
| Treatment | Isoprinosine + intraventricular IFN-α (no cure) |
| Prevention | MMR vaccination |
Sources:
- Bradley & Daroff's Neurology in Clinical Practice (primary)
- Harrison's Principles of Internal Medicine 22E (2025)
- Adams & Victor's Principles of Neurology 12E
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Goldman-Cecil Medicine (International Ed.)
Recent evidence note (PubMed 2024): A systematic review (Garg et al., 2024, PMID 38282559) documents the spectrum of psychiatric manifestations in SSPE - confirming that psychiatric features (personality change, mood disturbance) at Stage I are common presenting complaints and may delay diagnosis.
Please give full answer
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
"subacute sclerosing panencephalitis" MRI radiological
Reading Article
PMID: 42113281
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
I now have all the information needed from 7 standard textbooks plus the latest 2026 systematic review on MRI. Here is the full, comprehensive answer:
Subacute Sclerosing Panencephalitis (SSPE) - Complete Review
1. DEFINITION
Subacute sclerosing panencephalitis (SSPE) is a rare, uniformly fatal, chronic progressive demyelinating disease of the CNS caused by a persistent non-permissive (non-productive) infection of neurons and glial cells with wild-type measles virus. It develops years after the initial measles infection and is characterized by progressive intellectual deterioration, myoclonus, seizures, and eventual death.
(Harrison's 22E; Goldman-Cecil Medicine; Sherris Medical Microbiology)
2. ETIOLOGY AND PATHOGENESIS
Causative Agent
- Measles virus (Paramyxoviridae family, genus Morbillivirus) - a single-stranded, negative-sense RNA virus
- The virus responsible is wild-type (not vaccine strain); vaccination is protective
Why Does the Virus Persist?
The virus undergoes specific molecular defects in neural tissue that prevent infectious particle formation:
| Defect | Effect |
|---|---|
| Absent/mutated M (matrix) protein | Cannot assemble viral envelope; antibody to M protein is absent in patients |
| Restricted envelope protein expression | No budding/release of new virions |
| Reduced transcription efficiency in differentiated brain cells | Maintains persistence in intracellular form |
| Cell-to-cell spread (not humoral) | Hidden from immune surveillance despite very high antibody titers |
Key: The virus is concealed from the immune system inside cells. Despite the host mounting massive antimeasles antibody responses (highest measured antibody titers in any known condition), the virus cannot be cleared.
(Jawetz Medical Microbiology 28E; Sherris Medical Microbiology 8E; Bradley & Daroff's Neurology)
Epidemiology
- Incidence: ~1/100,000-500,000 measles cases; 4-11 per 100,000 measles survivors; 0.1-5 per million in unimmunized populations
- ~5 cases/year reported in the USA currently; several times higher in resource-limited countries
- Age at diagnosis: 85% between 5-15 years old
- Sex: Males predominate - male:female ratio = 3:1 (consistently across case series)
- Latent interval: Median 6-8 years after primary measles (range 2-12 years; Goldman-Cecil states 7-10 years)
- Primary infection age: Most had measles before age 2 years (key risk factor - early-life infection before full immune maturation)
- Incidence has declined dramatically with measles vaccination but may rise again with vaccine hesitancy and measles resurgence
3. CLINICAL FEATURES
There are no typical signs of acute CNS viral infection (no fever, no headache, no meningismus) - this distinguishes SSPE from acute encephalitis.
Jabbour Staging System (4 Stages)
Stage I - Behavioral / Mental Changes ("Insidious Stage")
Duration: Weeks to months
Features:
- Insidious onset of personality change - irritability, temper outbursts, emotional lability
- Decline in school performance - difficulty concentrating, memory lapses
- Difficulty with language - word-finding problems
- Social withdrawal, loss of interest in usual activities
- No myoclonus, no seizures at this stage
- Cognitive symptoms mimic psychiatric illness (depression, behavioral disorder)
- Often misdiagnosed at this stage - psychiatric referral is common
Mnemonic: "Bad Student" stage - behavioral + school decline
Stage II - Neurological / Motor Complications
Duration: Weeks to months
Features:
- Myoclonus - characteristic repetitive, shock-like muscle jerks; often symmetrical; may be triggered by sensory stimuli in advanced Stage II; this symptom typically brings the patient to medical attention
- Seizures - focal and/or generalized; may be refractory
- Progressive intellectual deterioration - severe dementia
- Ataxia - cerebellar gait disturbance
- Choreoathetoid or ballistic movements, extrapyramidal signs
- Chorioretinitis - visual disturbances; macular lesion appears white/grey-white, resolves to leave scarring and irregular RPE atrophy
- Papilloedema, nystagmus, optic neuritis may occur
- Spasticity develops, hyperactive deep tendon reflexes
- Posterior uveitis is common and may be the presenting feature (before obvious CNS involvement)
- The EEG periodic complexes become evident at this stage (synchronous with myoclonic jerks)
The classic Rademecker complexes on EEG are most prominent in Stage II
Stage III - Advanced / Pre-terminal ("Coma Stage")
Duration: Weeks to months
Features:
- Akinetic mutism - patient awake but completely unresponsive
- Quadriparesis
- Progressive unresponsiveness
- Optic atrophy - from prior chorioretinitis/optic neuritis
- Autonomic instability - thermoregulatory dysfunction, hyperhidrosis
- Dysphagia, incontinence
- Extensor plantar responses (Babinski sign)
- Acute episodes with raised intracranial pressure - poor prognostic sign
Stage IV - Vegetative / Terminal State
Duration: Weeks to months before death
Features:
- Child lies insensate - virtually "decorticated" or "decerebrate"
- Complete loss of cortical function
- Progressive inanition
- Superinfection (aspiration pneumonia, UTI, decubitus ulcers)
- Metabolic imbalances
- Death - within 1-3 years of onset in most cases
Prognosis
- Course is uniformly fatal in most cases
- Majority die within 1-3 years of symptom onset
- Spontaneous remissions: estimated ~5% (usually incomplete and temporary)
- Adult-onset SSPE: older patients (one series mean age 21 years, oldest 43); visual disturbances and extrapyramidal features more prominent, raising concern for prion disease; myoclonus present early
Ophthalmic Features (from Kanski's Ophthalmology 10E)
- Posterior uveitis may be the presenting feature before CNS symptoms
- Chorioretinitis with macular involvement - white/grey-white macular lesion
- Ischaemic retinal vein occlusion secondary to vasculitis (rare)
- Papilloedema, optic neuritis, nystagmus
- Congenital measles: cataract, retinopathy possible
4. STAGES - SUMMARY TABLE
| Stage | Duration | Key Features | EEG |
|---|---|---|---|
| I | Weeks-months | Behavioral change, school decline, personality changes | Nonspecific slowing |
| II | Weeks-months | Myoclonus, seizures, intellectual deterioration, chorioretinitis | Periodic complexes (Rademecker) |
| III | Weeks-months | Akinetic mutism, quadriparesis, autonomic instability | Suppression-burst; attenuated background |
| IV | Weeks-months | Vegetative state, decorticate/decerebrate posture, death | Nearly flat/isoelectric |
5. INVESTIGATIONS AND DIAGNOSTIC FEATURES
A. EEG - Pathognomonic Pattern ("Rademecker Complexes")
- Periodic bursts of high-voltage, sharp, slow-wave complexes (2-3 Hz), lasting up to 3 seconds
- Occur at regular intervals of 4-14 seconds (typically every 5-8 seconds)
- Against a background of depressed/attenuated ("flat") activity
- Synchronous with myoclonic jerks - this correlation is virtually diagnostic
- Early in disease: only nonspecific slowing
- Stage II: classic periodic pattern fully developed
- Late/terminal: nearly isoelectric
B. CSF Analysis
| Parameter | Finding |
|---|---|
| Cells | Acellular (no pleocytosis - distinguishes from acute encephalitis) |
| Glucose | Normal |
| Protein | Normal or mildly elevated |
| Gamma globulin | Markedly elevated (>20% of total CSF protein) |
| Oligoclonal bands | Present (measles-virus-specific IgG) |
| Antimeasles antibody | Invariably elevated - CSF/serum ratio confirms high intrathecal synthesis |
C. Serum
- Very high antimeasles antibody titers (among the highest ever measured in any infectious disease)
- Antibody to M protein frequently absent (marker of defective virus)
D. Virological Confirmation
- Measles virus can be cultured from brain tissue using special cocultivation techniques
- Immunocytochemistry: viral antigen in brain tissue
- In situ hybridization or PCR: viral genome detection in brain or CSF
- Brain biopsy: Cowdry type A inclusions + viral antigen
E. Diagnostic Criteria (Clinical)
In a child with characteristic staging, the following triad is sufficient for diagnosis (Adams & Victor):
- Periodic EEG complexes (Rademecker complexes)
- Elevated CSF gamma globulin + oligoclonal bands
- Elevated measles antibody titers in serum and CSF
6. RADIOLOGICAL FEATURES
MRI (Investigation of Choice)
The 2026 systematic review by Garg et al. (Neuroradiology, 2026; PMID 42113281) analyzed 461 confirmed SSPE cases across imaging studies - the most comprehensive neuroimaging data available:
Distribution of Findings (461 cases):
| Finding | Frequency |
|---|---|
| White matter abnormalities (total) | 93.5% |
| - Diffuse bilateral white matter | 28.6% |
| - Periventricular white matter | 22.6% |
| - Subcortical white matter | 15.4% |
| - Posterior parieto-occipital predominant | 13.7% |
| Cortical involvement | 53.6% |
| - Diffuse hemispheric | 16.1% |
| - Posterior predominance | 14.8% |
| Deep gray matter (basal ganglia/thalamus) | 17.2% |
| - Basal ganglia | 10.2% |
| - Thalamus | 5.0% |
| Brainstem abnormalities | 7.4% |
| Diffusion restriction (DWI) | 11.3% |
| Contrast enhancement | 8.2% (limited) |
| Cerebral atrophy | 35.1% (progressive atrophy in 13.7%) |
Stage-wise MRI Evolution:
Early (Stage I/Early Stage II):
- Often normal
- First changes: T2/FLAIR hyperintensities in subcortical white matter of posterior hemispheres (parieto-occipital region)
- Asymmetric, patchy
Established (Stage II):
- T2/FLAIR lesions extend anteriorly
- Cortical and subcortical involvement in temporal, parietal, frontal lobes
- Periventricular white matter involvement develops
- No/minimal contrast enhancement (BBB relatively intact)
Advanced (Stage III-IV):
- Lesions spread to involve brainstem
- Deep gray matter lesions - basal ganglia, thalamus
- Progressive cerebral atrophy - cortical and subcortical
- Confluent white matter changes resembling leukodystrophy
- Diffusion restriction may be seen (cytotoxic edema in acute active lesions)
Key MRI Characteristics:
- Posterior (parieto-occipital) > anterior distribution early
- White matter > gray matter early; both involved later
- No/minimal contrast enhancement (distinguishes from other encephalitides)
- Diffusion restriction when present = active demyelination/cytotoxic edema
- Progressive atrophy on serial imaging
- Cerebellum usually spared (classic teaching from Adams & Victor)
Radiological Mimics (Garg 2026):
| Pattern | % Cases |
|---|---|
| Ocular/visual pathway-predominant | 10.0% |
| Autoimmune encephalitis/ADEM-like | 8.0% |
| Leukodystrophy-like | 6.7% |
| Stroke-like | 6.3% |
| Basal ganglia-predominant | 5.6% |
CT Scan
- Less sensitive than MRI
- May be normal early
- Later: low-density white matter lesions, cerebral atrophy
- CT used in 12.6% of cases in systematic review (mostly resource-limited settings)
7. PATHOLOGY (Histopathology)
Macroscopic
- Brain atrophy, firm white matter (sclerosing)
- Grey and white matter of both cerebral hemispheres and brainstem affected
- Cerebellum usually spared
Microscopic (H&E)
- Cowdry type A eosinophilic intranuclear inclusions in neurons and glial cells - hallmark of SSPE
- Cigar-shaped cytoplasmic inclusions in neurons
- Destruction of nerve cells, neuronophagia
- Perivenous cuffing by lymphocytes and mononuclear cells
- White matter: demyelination of axons and myelin sheaths
- Fibrous gliosis (hence "sclerosing")
- Both gray matter (cortex) and white matter involved
Electron Microscopy
- Measles nucleocapsids visible within inclusion-bearing cells
8. DIFFERENTIAL DIAGNOSIS
| Condition | Distinguishing Features |
|---|---|
| Creutzfeldt-Jakob disease | Older age; spongiform change on biopsy; 14-3-3 protein in CSF; no measles antibody elevation |
| Lipid storage diseases | Specific enzyme deficiencies; different histology |
| Schilder's demyelinating disease | Demyelination pattern; no measles antibodies |
| Childhood dementing illnesses | No periodic EEG; no measles antibody elevation |
| Progressive rubella panencephalitis | Older patients; more protracted course; lacks generalized myoclonus and periodic EEG bursts |
| Autoimmune encephalitis | Anti-neuronal antibodies; responds to immunotherapy |
| ADEM | Monophasic; post-infectious; perivenous demyelination |
9. TREATMENT
No curative treatment exists. The goal is to slow progression and provide symptomatic relief.
A. Antiviral/Immunomodulatory Therapy
| Drug | Dose | Route | Notes |
|---|---|---|---|
| Isoprinosine (Inosiplex / Inosine pranobex) | 100 mg/kg/day (max 3 g/day) in 3 divided doses | Oral | Most widely used; reported to prolong survival and produce improvement in some; never tested in controlled RCT; used for 6 months |
| Intraventricular Interferon-α (via Ommaya reservoir) | Start 100,000 U/m² BSA/day → escalate to 10⁶ U/m²/day over 5 days → then 10⁶ U/m² twice weekly for 6 months | Intraventricular | ~30-35% of patients improve or stabilize; risks: meningitis, IFN-encephalopathy, motor neuron toxicity |
| Combined Isoprinosine + Intraventricular IFN-α | As above for both | Oral + intraventricular | Best response reported; ~30-35% improved/stabilized (Gascon 2003; Gutierrez 2010) |
| Subcutaneous Interferon-α | Up to 5 million U/day | SC | Used alongside intrathecal IFN to treat peripheral measles reservoirs (lymphoid, glandular tissue) |
| Intrathecal Interferon-α | Variable | Intrathecal | Alternative to intraventricular; less well established |
| Ribavirin (IV) + intrathecal IFN-α | Variable | IV | Case reports only; some benefit (Tomoda et al. 2001) |
| Amantadine | Oral | Oral | Historical use; improvement not consistently confirmed (Adams & Victor) |
B. Symptomatic Treatment
| Symptom | Treatment |
|---|---|
| Myoclonus / Seizures | Levetiracetam (shown to improve myoclonus and encephalopathy); other antiepileptics (valproate, clonazepam, clobazam) |
| Spasticity | Baclofen, physiotherapy |
| Autonomic dysfunction | Symptomatic management |
| Nutritional support | NG tube / PEG feeding as disease progresses |
| Respiratory support | Ventilatory support in terminal stages |
| Superinfection | Antibiotics for pneumonia/UTI |
C. Treatment Monitoring
- Laboratory endpoint: Eradication of detectable measles antigen from CSF
- Serial clinical assessment and EEG
- Risks of prolonged IFN therapy: meningitis, IFN-induced encephalopathy, upper and lower motor neuron toxicity
D. Spontaneous Remission
- Approximately 5% of cases show spontaneous (usually temporary) improvement - always consider when evaluating treatment response
10. PREVENTION
- MMR vaccine (Measles-Mumps-Rubella) - most important intervention
- First dose: 12-15 months of age
- Second dose: 4-6 years of age
- Also available as MMRV (quadrivalent: measles, mumps, rubella, varicella)
- Measles vaccination has virtually eliminated SSPE in countries with high coverage
- Rising vaccine hesitancy and measles resurgence (including in the USA) may cause an increase in SSPE cases in the coming decades (Harrison's 22E warning)
- Post-exposure prophylaxis with immunoglobulin within 6 days of measles exposure (especially for children <1 year, pregnant women, immunocompromised)
11. COMPLETE DIAGNOSTIC CRITERIA SUMMARY
A clinical diagnosis of SSPE can be made when the following are present (Adams & Victor):
- Characteristic clinical picture - Progressive neurological deterioration in a child with history of early measles
- Periodic EEG - High-voltage slow-wave complexes every 5-8 seconds (Rademecker complexes)
- CSF: Elevated gamma globulin (>20%), oligoclonal bands, acellular
- Serum and CSF antimeasles antibody: Invariably elevated
- Confirmatory (if needed): Brain biopsy - Cowdry A inclusions; PCR for measles genome; viral antigen by immunocytochemistry; viral culture by cocultivation
12. OVERVIEW TABLE
| Feature | Details |
|---|---|
| Cause | Persistent wild-type measles virus (defective, non-productive CNS infection) |
| Mechanism | Absent M protein → no viral budding → intracellular spread only → immune evasion |
| Incidence | 4-11/100,000 measles survivors; ~5 cases/year in USA |
| Peak age | 5-15 years (85% of cases) |
| Sex | Male:Female = 3:1 |
| Latency | 6-8 years after primary measles (range 2-12 years) |
| Risk factor | Measles before age 2 years |
| Stage I | Behavioral change, school decline (no myoclonus) |
| Stage II | Myoclonus, seizures, chorioretinitis, intellectual deterioration |
| Stage III | Akinetic mutism, quadriparesis, autonomic instability |
| Stage IV | Vegetative state, death |
| EEG hallmark | Periodic high-voltage slow waves every 3-14 sec (Rademecker complexes) |
| CSF hallmark | Acellular; markedly elevated gamma globulin; antimeasles antibody; oligoclonal bands |
| MRI | T2/FLAIR: posterior subcortical → white matter → diffuse; no/minimal enhancement; atrophy late |
| Histology hallmark | Cowdry type A intranuclear inclusions; cigar-shaped cytoplasmic inclusions |
| Treatment | Isoprinosine + intraventricular IFN-α (no cure); levetiracetam for myoclonus |
| Prognosis | Uniformly fatal; 1-3 years survival; 5% spontaneous remission |
| Prevention | MMR vaccine (virtually eliminates SSPE) |
Sources (Standard Textbooks):
- Bradley & Daroff's Neurology in Clinical Practice - primary neurology reference
- Harrison's Principles of Internal Medicine 22E (2025, McGraw Hill)
- Adams & Victor's Principles of Neurology 12E
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Goldman-Cecil Medicine (International Edition)
- Jawetz, Melnick & Adelberg's Medical Microbiology 28E
- Sherris & Ryan's Medical Microbiology 8E
- Kanski's Clinical Ophthalmology: A Systematic Approach 10E
Recent Evidence:
- Garg RK et al. "The neuroimaging spectrum of SSPE: a systematic review." Neuroradiology 2026 (PMID 42113281) - comprehensive MRI data from 461 cases confirming predominant posterior bilateral white matter involvement with stage-evolving pattern
- Garg RK et al. "Psychiatric manifestations in SSPE." CNS Spectr 2024 (PMID 38282559) - confirms psychiatric features are common at Stage I and frequently delay diagnosis
This is a shared conversation. Sign in to Orris to start your own chat.