AML , ALL , CML , CLL DIFFERENT AND DIFFERENTIAL DIAGNOSIS.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive textbook data and histological images to produce a thorough, well-organized answer. Here it is:
Leukemias: AML, ALL, CML, CLL - Comparison and Differential Diagnosis
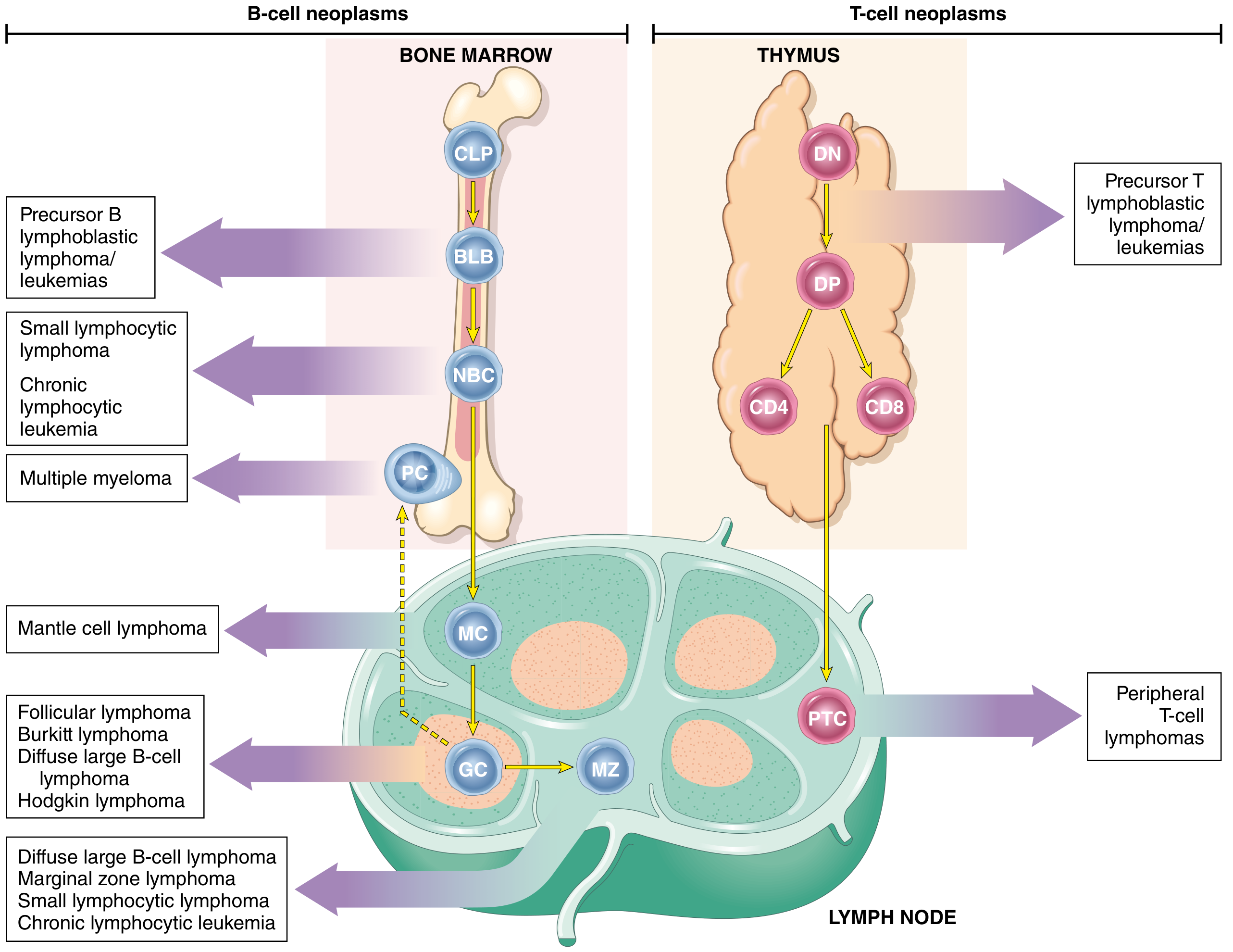
Origin of Lymphoid vs. Myeloid Neoplasms
Overview Table: Four Leukemias at a Glance
| Feature | AML | ALL | CML | CLL |
|---|---|---|---|---|
| Cell of origin | Myeloid progenitor | Lymphoid precursor (B or T) | Myeloid stem cell | Mature B lymphocyte |
| Course | Acute | Acute | Chronic (3 phases) | Chronic (indolent) |
| Peak age | >60 years | Children (peak 3 yrs) | 45-55 years | >60 years |
| Sex | M=F | M slightly > F | M slightly > F | M:F = 2:1 |
| Key genetic marker | t(15;17), t(8;21), inv(16), NPM1, FLT3 | BCR::ABL1 (Ph+), hyperdiploidy, NOTCH1 | t(9;22) Philadelphia chromosome (BCR::ABL1) | del 13q14, trisomy 12, del 11q, del 17p |
| WBC count | Variable (elevated or low) | Variable (elevated, normal, or low) | Markedly elevated (50,000-200,000/µL) | Markedly elevated (small lymphocytes) |
| Blast count (BM) | ≥20% myeloblasts | ≥20% lymphoblasts | <10% (chronic); >20% (blast crisis) | Rare blasts; mature lymphocytes |
| Auer rods | Present (pathognomonic) | Absent | Absent | Absent |
| Splenomegaly | Mild/absent | Mild | Massive | Moderate |
| Lymphadenopathy | Mild | Common | Mild | Diffuse (prominent) |
| Key treatment | Cytarabine + anthracycline; ATRA for APL | Multiagent chemo; TKI for Ph+ | Imatinib (TKI) | Ibrutinib, FCR, venetoclax |
| Prognosis | Variable; poor in elderly | 95% remission in children; worse in adults | Excellent with TKI; depends on phase | Indolent; incurable but long survival |
1. Acute Myeloid Leukemia (AML)
Definition & Epidemiology
AML is a tumor of hematopoietic progenitors caused by acquired oncogenic mutations that impede differentiation, leading to accumulation of immature myeloid blasts in the marrow. It is the most common acute leukemia in adults, with median age of 60 years (incidence rises to 10/100,000/year in those >60). About 13,000 new US cases/year.
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Clinical Features
- Onset resembles acute infection: fever, ulcerations of mucous membranes (mouth/throat), fatigue, pallor
- Granulocytic insufficiency: infections, mouth ulcers
- Anemia and thrombocytopenia (marrow failure)
- Lymphadenopathy, splenomegaly, and hepatomegaly are NOT pronounced (distinguishes from CML/ALL)
- Untreated: rapidly progressive
Risk Factors
- Prior exposure to benzene, ionizing radiation, cytotoxic chemotherapy
- Congenital syndromes: Down syndrome, Fanconi anemia, neurofibromatosis, Noonan syndrome
Morphology & Diagnosis
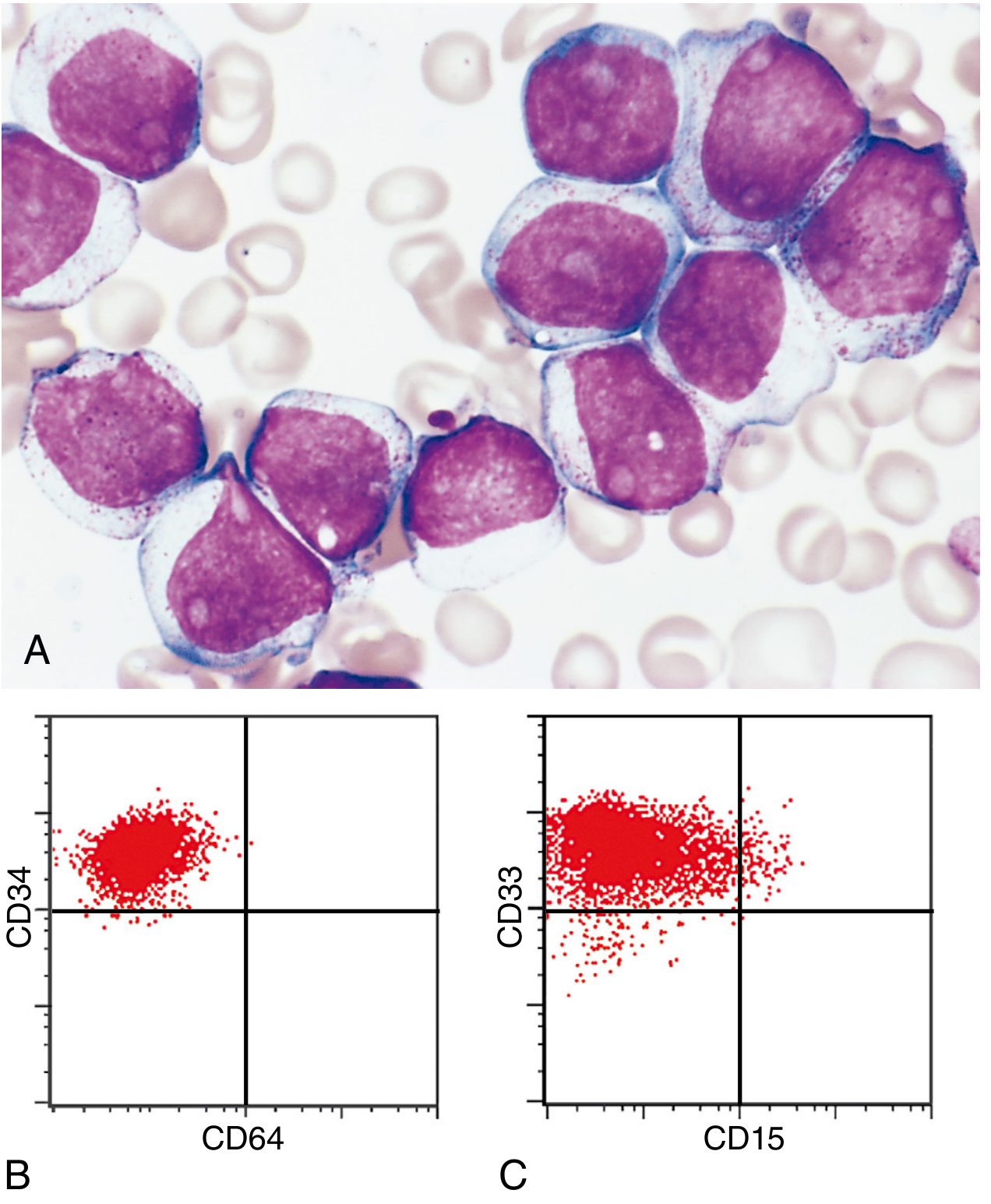
- Diagnosis: ≥20% blasts in bone marrow or blood
- Auer rods (eosinophilic needle-like cytoplasmic inclusions from MPO-positive granules): pathognomonic for AML, absent in ALL
- Myeloblasts: 2-4 nucleoli, voluminous cytoplasm, peroxidase-positive granules
- Cytochemistry: MPO+, Sudan Black B+, chloroacetate esterase+
Key Genetic Subtypes (WHO Classification)
| Genetic Abnormality | Prognosis | Notes |
|---|---|---|
| t(8;21); RUNX1::RUNX1T1 | Favorable | Core binding factor AML; Auer rods easily found |
| inv(16); CBFB::MYH11 | Favorable | Myelocytic + monocytic differentiation |
| t(15;17); PML::RARA | Very favorable | APL - responds to ATRA; numerous Auer rods; high DIC risk |
| KMT2A (11q23) rearrangement | Poor | Monocytic differentiation; gingival hypertrophy |
| Mutated NPM1 | Favorable | Most common single mutation in AML |
| Myelodysplasia-related genetics | Poor | del 5q/7q; SF3B1, SRSF2 mutations |
| TP53 mutations | Very poor | Complex karyotype, resistance to standard therapy |
Immunophenotype
CD13+, CD33+, CD117+, MPO+, CD34+ (variable); CD3-, CD19-, CD20- (negative for lymphoid markers)
2. Acute Lymphoblastic Leukemia (ALL)
Definition & Epidemiology
ALL is composed of immature B (pre-B) or T (pre-T) cells (lymphoblasts). It is the most common cancer of childhood (~2,500 new cases/year in the US). About 85% are B-ALL; T-ALL tends to present in adolescent males as thymic lymphomas.
- Peak incidence: 2-3 years of age for B-ALL; adolescence for T-ALL
- Slightly more frequent in boys; highest incidence in Hispanic/Latino children
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Clinical Features
- Intermittent fevers, bone pain, pallor
- Cutaneous/mucosal bleeding, petechiae, purpura
- Lymphadenopathy and hepatosplenomegaly: prominent (unlike AML)
- Testicular enlargement, subcutaneous nodules (leukemia cutis)
- Mediastinal widening on CXR (T-ALL - anterior mediastinal mass)
- WBC: elevated, normal, or low; + neutropenia, anemia, thrombocytopenia
Molecular Pathogenesis
- T-ALL: NOTCH1 mutations (essential for T-cell development)
- B-ALL: mutations in PAX5, TCF3, ETV6, RUNX1, BCR::ABL1, KMT2A, PBX1
- Chromosomal: ~90% have numerical/structural changes
- Hyperdiploidy (>50 chr): B-ALL, better prognosis
- Hypodiploidy: B-ALL, worse prognosis
- t(9;22) BCR::ABL1 (Philadelphia chromosome): 25% of adult ALL, poor prognosis - requires TKI
- t(12;21) ETV6::RUNX1: most common translocation in childhood B-ALL, excellent prognosis
Morphology & Diagnosis
- Lymphoblasts: scant cytoplasm, finer chromatin, less cytoplasm than myeloblasts
- No Auer rods
- Diagnosis: ≥20% lymphoblasts in BM
Immunophenotype
- B-ALL: CD19+, CD10+ (CALLA), CD22+, TdT+, CD34+, MPO-
- T-ALL: CD3+, CD7+, TdT+, CD34+, MPO-
- Both: MPO negative (key distinction from AML)
Prognosis
- Children: 95% remission rate; ~80% disease-free at 5 years
- Adults: significantly worse outcomes
- Ph+ ALL requires TKI (imatinib/dasatinib) added to chemotherapy
3. Chronic Myeloid Leukemia (CML)
Definition & Epidemiology
CML is defined by the Philadelphia chromosome - a reciprocal translocation t(9;22)(q34;q11) producing the BCR::ABL1 fusion gene. This generates a constitutively active tyrosine kinase (p210 fusion protein) that drives uncontrolled myeloid proliferation.
CML Histology (peripheral blood and bone marrow):
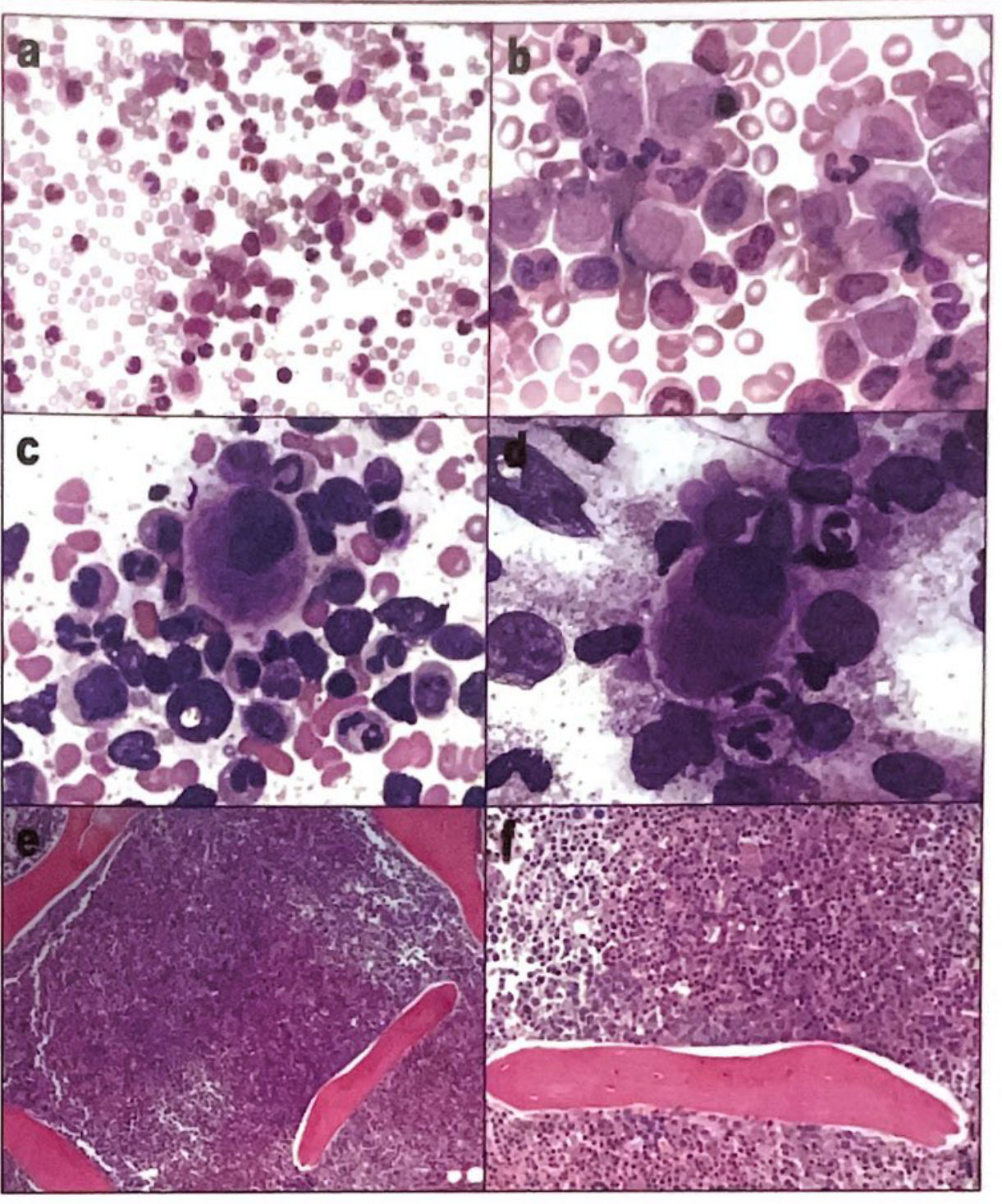
Three Phases of CML
Chronic Phase
- Leukocytosis (50,000-200,000/µL): neutrophils in all stages of maturation
- Myelocyte "bulge" (proportion of myelocytes exceeds other immature forms)
- Basophilia + eosinophilia + thrombocytosis (pathognomonic combination)
- Low LAP (leukocyte alkaline phosphatase) score - key distinguishing finding from leukemoid reaction
- Blasts: usually <1%
- Absolute monocytosis in most cases
- Massive splenomegaly
Accelerated Phase
One or more of:
- Progressive basophilia (>20%)
- Thrombocytopenia (<100 × 10⁹/L) unrelated to therapy
- Leukocytosis unresponsive to therapy
- Clonal cytogenetic progression (extra Ph chromosome, +8, i17q, +19)
- Blasts 10-19%
Blast Phase (Blast Crisis)
- >20% blasts in blood or marrow - becomes acute leukemia
- 70% are AML type, 30% are ALL type
- Additional cytogenetic abnormalities
- Poorly responsive to treatment
Key Lab Findings
| Test | CML | Leukemoid Reaction |
|---|---|---|
| LAP score | Low | High |
| Philadelphia chromosome | Positive | Negative |
| BCR::ABL1 by PCR | Positive | Negative |
| Basophilia | Present | Absent |
Treatment
- Imatinib (Gleevec) - competitive inhibitor of ATP binding site of BCR::ABL; also active against PDGFR and c-KIT
- 2nd line: nilotinib, dasatinib
- Resistance mechanism: T315I mutation in BCR::ABL kinase domain; MDR1 overexpression; BCR::ABL amplification
- Prognostic monitoring: quantitative RT-PCR for BCR::ABL1 transcript (MRD)
4. Chronic Lymphocytic Leukemia (CLL)
Definition & Epidemiology
CLL is a clonal proliferation of small mature B lymphocytes involving bone marrow, blood, and lymph nodes. It is the most common chronic lymphoid leukemia in the US and Europe (90% of cases). CLL and Small Lymphocytic Lymphoma (SLL) are considered the same entity - CLL when predominantly leukemic, SLL when predominantly nodal.
- Rare below 40 years; most cases >60 years
- Male:Female ratio >2:1
- Onset insidious, commonly discovered incidentally
- Henry's Clinical Diagnosis and Management by Laboratory Methods
Clinical Features
- Painless lymphadenopathy (diffuse)
- Hepatosplenomegaly
- Fatigue, weight loss, recurrent infections (hypogammaglobulinemia)
- Autoimmune hemolytic anemia or ITP in 10-25%
- WBC: markedly elevated, predominantly small mature lymphocytes with "smudge cells" on blood smear
Morphology
- Diffuse infiltrates of small mature lymphocytes with clumped chromatin and scant cytoplasm
- Admixed prolymphocytes and proliferation centers (pseudofollicles) - characteristic
- Mitotic rates: very low
- Smudge (smear) cells on peripheral smear: pathognomonic
Immunophenotype (Critical for Diagnosis)
- B-cell markers: CD19+, CD20+(dim), CD79a+
- Dim surface Ig (monoclonal IgM or IgM + IgD)
- CD5+ (T-cell marker aberrantly expressed: key feature distinguishing from other B-cell lymphomas)
- CD23+, CD200+, LEF1+
- CD10-, BCL6-, Cyclin D1- (distinguishes from follicular and mantle cell lymphomas)
Cytogenetics & Prognostic Markers
| Abnormality | Frequency | Prognosis |
|---|---|---|
| del(13q14.3) | 50% | Favorable |
| Trisomy 12 | 20% | Intermediate |
| del(11q22-23) (ATM) | 20% | Adverse |
| del(17p13) (TP53) | 10% | Adverse |
| del(6q21) | 5% | Adverse |
- IGHV mutation status: Mutated IGHV = better prognosis; Unmutated IGHV (ZAP-70+) = adverse prognosis
- Other adverse factors: CD38+, CD49d+, lymphocyte doubling time <12 months, elevated β₂-microglobulin
Differential Diagnosis Between the Four Leukemias
Step-by-Step Approach
Step 1: Acute vs. Chronic
| Acute (AML/ALL) | Chronic (CML/CLL) | |
|---|---|---|
| Blasts in BM | ≥20% | <10% (CML chronic phase) or absent (CLL) |
| Onset | Abrupt, weeks | Insidious, months-years |
| WBC differential | Blasts predominate | Mature/maturing cells predominate |
| Clinical urgency | Medical emergency | Often incidental finding |
Step 2: Myeloid vs. Lymphoid
| Feature | Myeloid (AML, CML) | Lymphoid (ALL, CLL) |
|---|---|---|
| Auer rods | Present (AML) | Absent |
| MPO/Sudan Black | Positive | Negative |
| Cytochemistry | NSE+ (monocytic AML) | Negative |
| Flow cytometry | CD13, CD33, CD117, MPO | CD19, CD10, CD3, TdT |
| LAP score | Low (CML) | Normal/variable |
Step 3: Within acute leukemias (AML vs. ALL)
| AML | ALL | |
|---|---|---|
| Auer rods | Present (pathognomonic) | Absent |
| Cell size | Larger; more cytoplasm | Smaller; scant cytoplasm |
| Nucleoli | 2-4, prominent | 1-2, less prominent |
| MPO | Positive | Negative |
| TdT | Usually negative | Positive |
| CD markers | CD13, CD33, CD117, CD34 | CD19/CD10 (B-ALL); CD3/CD7 (T-ALL) |
| Age preference | Adults (>60 yrs) | Children (peak 2-3 yrs) |
| Mediastinal mass | Absent | T-ALL only |
Step 4: Within chronic leukemias (CML vs. CLL)
| CML | CLL | |
|---|---|---|
| Cell type | Myeloid (neutrophils + precursors) | Lymphoid (small B lymphocytes) |
| Philadelphia chromosome | Positive | Negative |
| BCR::ABL1 | Positive | Negative |
| LAP score | Low | Normal |
| Basophilia | Prominent | Absent |
| Smudge cells | Absent | Present |
| CD5+ B cells | No | Yes |
| Splenomegaly | Massive | Moderate |
| Lymphadenopathy | Mild | Prominent |
| Auer rods | Absent (unless blast crisis) | Absent |
Differential Diagnosis from Other Conditions
AML vs. Leukemoid Reaction
| AML | Leukemoid Reaction | |
|---|---|---|
| Auer rods | Present | Absent |
| LAP score | Variable (low in CML) | High |
| Blast count | ≥20% | <5% |
| Philadelphia chr | Negative | Negative |
| Cause | Neoplastic | Infection/inflammation |
CLL vs. Other Small B-Cell Lymphomas
| CLL/SLL | Mantle Cell | Follicular | Marginal Zone | |
|---|---|---|---|---|
| CD5 | + | + | - | - |
| CD23 | + | - | - | - |
| CD10 | - | - | + | - |
| Cyclin D1 | - | + | - | - |
| LEF1 | + | - | - | - |
| t(11;14) | Absent | Present | Absent | Absent |
ALL vs. Lymphoblastic Lymphoma
- Same disease - differentiated only by degree of BM involvement
- >25% blasts in BM = leukemia; <25% = lymphoma (by convention)
Quick Memory Mnemonics
- AML: Auer rods, Myeloperoxidase+, Middle-aged to elderly, Marrow failure presentation
- ALL: All children (most common childhood cancer), Absent Auer rods, Anterior mediastinal mass (T-ALL), ACALLA (CD10)+
- CML: Chromosome 9+22 (Philadelphia), Chronic course with 3 phases, Causes massive spleen, Cured by imatinib
- CLL: CD5+ B-cells, Clumped chromatin, Common in elderly men, Crushed smudge cells
Treatment Summary
| Leukemia | First-line | Notes |
|---|---|---|
| AML | Cytarabine + anthracycline ("7+3") | APL (M3): ATRA + arsenic trioxide; IDH inhibitors for mutated IDH1/2 |
| ALL | Multi-agent chemotherapy (CALGB/GRAALL protocols) | Ph+ ALL adds imatinib/dasatinib; CNS prophylaxis essential |
| CML | Imatinib (1st gen TKI) | Nilotinib/dasatinib (2nd gen) for resistance/intolerance; T315I: ponatinib |
| CLL | Ibrutinib (BTK inhibitor), venetoclax (BCL-2 inhibitor), FCR | Watch-and-wait for early-stage; del(17p)/TP53 mutation requires ibrutinib/venetoclax |
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528)
- Henry's Clinical Diagnosis and Management by Laboratory Methods (9780323673204)
- Tietz Textbook of Laboratory Medicine, 7th Edition (9780323775724)
- Quick Compendium of Clinical Pathology, 5th Edition (9780891896852)
- Swanson's Family Medicine Review (9780323698115)
- Goldman-Cecil Medicine (9780323930345)
AML , ALL , CML , CLL DIFFERENT AND DIFFERENTIAL DIAGNOSIS.
The comprehensive answer I just provided covers exactly this topic in full detail. Here is a complete summary for quick reference:
AML · ALL · CML · CLL — Differences & Differential Diagnosis
1. Quick Orientation: The 2×2 Framework
ACUTE CHRONIC
MYELOID AML (blasts ≥20%) CML (Ph chromosome)
LYMPHOID ALL (blasts ≥20%) CLL (mature B cells)
2. Individual Disease Profiles
AML — Acute Myeloid Leukemia
| Feature | Detail |
|---|---|
| Cell of origin | Myeloid progenitor (maturation arrested) |
| Age | Adults; median age 60 years (most common acute leukemia in adults) |
| Onset | Abrupt - days to weeks |
| Presentation | Fever, mouth ulcers (granulocyte failure), pallor, bleeding; spleen/lymph nodes NOT prominently enlarged |
| WBC | Variable - can be high, normal, or low |
| Blast count | ≥20% myeloblasts in BM/blood |
| Pathognomonic finding | Auer rods (eosinophilic needle-like cytoplasmic inclusions) |
| Cytochemistry | MPO+, Sudan Black B+, chloroacetate esterase+ |
| Immunophenotype | CD13+, CD33+, CD117+, MPO+, TdT- (usually) |
| Key genetics | t(15;17) APL = ATRA-sensitive; t(8;21), inv(16) = favorable; NPM1 mutation = favorable; FLT3-ITD, TP53 = poor |
| DIC risk | APL [t(15;17)] - hematologic emergency |
| Treatment | Cytarabine + anthracycline ("7+3"); ATRA + arsenic for APL |
| Prognosis | Variable; ~70% remission in fit patients; very poor in elderly |
ALL — Acute Lymphoblastic Leukemia
| Feature | Detail |
|---|---|
| Cell of origin | Immature B-cell precursor (85%) or T-cell precursor (15%) |
| Age | Most common cancer in children (peak 2-3 yrs); also occurs in adults (worse prognosis) |
| Onset | Abrupt |
| Presentation | Fever, bone pain, pallor, prominent lymphadenopathy + hepatosplenomegaly; T-ALL → anterior mediastinal mass; testicular involvement possible |
| WBC | Variable - elevated, normal, or low + neutropenia, anemia, thrombocytopenia |
| Blast count | ≥20% lymphoblasts in BM |
| Auer rods | Absent (critical distinction from AML) |
| Cytochemistry | MPO negative; PAS+ (B-ALL); acid phosphatase+ (T-ALL) |
| Immunophenotype | B-ALL: CD19+, CD10+ (CALLA), CD22+, TdT+, CD34+ |
| T-ALL: CD3+, CD7+, TdT+, CD34+ | |
| Key genetics | Hyperdiploidy (>50 chr) = good; Hypodiploidy = poor; t(9;22) Ph+ = poor (25% adult ALL); t(12;21) ETV6::RUNX1 = excellent (childhood); NOTCH1 (T-ALL) |
| CNS | CNS prophylaxis essential; CNS involvement common |
| Treatment | Multi-agent chemo; Ph+ ALL adds imatinib/dasatinib |
| Prognosis | Children: 95% remission, 80% cure; adults: ~40% cure |
CML — Chronic Myeloid Leukemia
| Feature | Detail |
|---|---|
| Cell of origin | Myeloid stem cell |
| Age | 45-55 years; gradual increase with age |
| Defining lesion | Philadelphia chromosome t(9;22)(q34;q11) → BCR::ABL1 fusion → constitutive tyrosine kinase activity (p210) |
| Onset | Insidious; often discovered incidentally |
| Presentation | Fatigue, massive splenomegaly, weight loss, night sweats; mild lymphadenopathy |
| WBC | Markedly elevated (50,000-200,000/µL) with full myeloid spectrum |
| Peripheral smear | All stages of myeloid maturation; myelocyte "bulge"; basophilia + eosinophilia + thrombocytosis |
| Blasts | <10% (chronic phase) |
| LAP score | LOW (key test - distinguishes from leukemoid reaction) |
| BM | Hypercellular; "dwarf" megakaryocytes; thickened paratrabecular cuff |
| Auer rods | Absent (unless blast crisis) |
Three Phases of CML
| Phase | Blasts | Key Features |
|---|---|---|
| Chronic | <10% | Manageable; responds well to TKI |
| Accelerated | 10-19% | Basophilia >20%; cytogenetic progression; thrombocytopenia |
| Blast Crisis | ≥20% | Becomes acute leukemia; 70% AML type, 30% ALL type |
| Treatment | Notes |
|---|---|
| Imatinib (1st gen TKI) | Competes at ATP binding site of BCR::ABL; also active vs PDGFR, c-KIT |
| Nilotinib, Dasatinib | 2nd generation; for resistance/intolerance |
| Ponatinib | For T315I "gatekeeper" mutation |
| MRD monitoring | Quantitative RT-PCR for BCR::ABL1 transcript |
CLL — Chronic Lymphocytic Leukemia
| Feature | Detail |
|---|---|
| Cell of origin | Mature monoclonal B-lymphocyte (naïve B-cell phenotype) |
| Age | >60 years; rare <40 years; M:F > 2:1 |
| Onset | Very insidious; often found on routine CBC |
| Presentation | Painless diffuse lymphadenopathy, hepatosplenomegaly, recurrent infections, fatigue; autoimmune hemolytic anemia in 10-25% |
| WBC | Markedly elevated - predominantly small mature lymphocytes |
| Peripheral smear | Monotonous small lymphocytes + smudge (smear) cells - pathognomonic |
| Blasts | Rare |
| Morphology | Small lymphocytes with clumped chromatin, scant cytoplasm; proliferation centers (pseudofollicles) in lymph nodes/BM |
CLL Immunophenotype (Critical for Diagnosis)
| Marker | CLL | Meaning |
|---|---|---|
| CD5+ | Positive | Aberrant T-cell marker on B-cells - KEY feature |
| CD23+ | Positive | Distinguishes from Mantle Cell |
| CD200+, LEF1+ | Positive | Distinguishes from other B-cell NHL |
| CD20+ | Dim | Weak expression |
| Surface Ig | Dim/monoclonal | IgM ± IgD |
| CD10- | Negative | Distinguishes from follicular lymphoma |
| Cyclin D1- | Negative | Distinguishes from mantle cell |
CLL Cytogenetic Prognostic Markers
| Abnormality | Frequency | Prognosis |
|---|---|---|
| del(13q14.3) | 50% | Favorable |
| Trisomy 12 | 20% | Intermediate |
| del(11q22-23) [ATM] | 20% | Adverse |
| del(17p13) [TP53] | 10% | Very adverse |
| del(6q21) | 5% | Adverse |
- IGHV unmutated + ZAP-70+ = aggressive course
- Treatment: ibrutinib (BTK inhibitor), venetoclax (BCL-2 inhibitor), FCR; watch-and-wait for Rai stage 0-II without symptoms
3. Side-by-Side Differential Diagnosis
Step 1 — Acute vs. Chronic (First split)
| Feature | Acute (AML/ALL) | Chronic (CML/CLL) |
|---|---|---|
| BM blasts | ≥20% | <10% or absent |
| Onset | Abrupt (days-weeks) | Insidious (months-years) |
| Emergency? | Yes | Rarely |
| Mature cells on smear | Few | Predominant |
| Symptoms at diagnosis | Severe | Often none |
Step 2 — Myeloid vs. Lymphoid (Second split)
| Feature | Myeloid (AML, CML) | Lymphoid (ALL, CLL) |
|---|---|---|
| Auer rods | AML: Present | Absent always |
| MPO / Sudan Black | Positive | Negative |
| TdT | Negative (usually) | Positive (ALL) |
| CD3/CD19/CD20 | Negative | Positive |
| CD13/CD33/CD117 | Positive | Negative |
| Philadelphia chr | CML: Positive | ALL: 25% adult cases |
| Basophilia | CML: prominent | Absent |
| Smudge cells | Absent | CLL: Present |
Step 3 — AML vs. ALL (Acute leukemias)
| Feature | AML | ALL |
|---|---|---|
| Auer rods | Present - pathognomonic | Absent |
| MPO | Positive | Negative |
| TdT | Negative | Positive |
| Cell size | Large; more cytoplasm | Small; scant cytoplasm |
| Nucleoli | 2-4, prominent | 1-2, less prominent |
| CD markers | CD13, CD33, CD117, CD34 | CD19/CD10 (B) or CD3/CD7 (T) |
| Age | Adults (>60 yrs) | Children (peak 2-3 yrs) |
| Mediastinal mass | Absent | T-ALL only |
| Hepatosplenomegaly | Mild | Prominent |
| Lymphadenopathy | Mild | Prominent |
| Gingival hypertrophy | AML-M5 (monocytic) | Absent |
Step 4 — CML vs. CLL (Chronic leukemias)
| Feature | CML | CLL |
|---|---|---|
| Cell type | Myeloid (all stages) | Mature B lymphocytes |
| Philadelphia chromosome | Positive | Negative |
| BCR::ABL1 PCR | Positive | Negative |
| LAP score | Low | Normal |
| Basophilia | Prominent | Absent |
| Smudge cells | Absent | Present |
| CD5+ B-cells | No | Yes |
| WBC morphology | Full myeloid spectrum | Monotonous small lymphocytes |
| Splenomegaly | Massive | Moderate |
| Lymphadenopathy | Mild | Diffuse, prominent |
| Auer rods | Absent | Absent |
| Thrombocytosis | Common | Absent (thrombocytopenia late) |
Step 5 — CML vs. Leukemoid Reaction
| Feature | CML | Leukemoid Reaction |
|---|---|---|
| LAP score | Low | High |
| Philadelphia chromosome | Positive | Negative |
| Basophilia | Present | Absent |
| Blasts | Present | Rare |
| Cause | Neoplastic | Infection/sepsis/inflammation |
| Toxic granulation | Absent | Present |
Step 6 — CLL vs. Other Small B-Cell Lymphomas
| Marker | CLL/SLL | Mantle Cell | Follicular | Marginal Zone |
|---|---|---|---|---|
| CD5 | + | + | - | - |
| CD23 | + | - | - | - |
| CD10 | - | - | + | - |
| Cyclin D1 | - | + | - | - |
| LEF1 | + | - | - | - |
| t(11;14) | Absent | Present | Absent | Absent |
| t(14;18) | Absent | Absent | Present | Absent |
4. Memory Mnemonics
| Leukemia | Mnemonic |
|---|---|
| AML | Auer rods + MPO+ + Mature patients + Marrow failure |
| ALL | All children + Absent Auer rods + Anterior mediastinum (T-ALL) + ACALLA (CD10+) |
| CML | Chromosome 22 (Ph) + Chronic 3-phase course + Colossal spleen + Cured by imatinib |
| CLL | CD5+ B-cells + Clumped chromatin + Crushed smudge cells + Common in old men |
5. Key Diagnostic Tests Ordered in Sequence
Suspected leukemia
↓
CBC + peripheral smear
↓
Is patient acute (sick, blasts on smear) or chronic (incidental)?
↓ ↓
ACUTE: BM aspirate CHRONIC: Flow cytometry
(blast count, morphology) (immunophenotype)
↓ ↓
MPO/TdT/flow cytometry Ph chromosome / BCR-ABL PCR
↓ ↓
AML if MPO+/Auer rods CML if Ph+ / BCR-ABL+
ALL if TdT+/MPO- CLL if CD5+CD23+B-cells
↓ ↓
Cytogenetics + molecular FISH for del13q, del17p
(prognostication + Rx) IGHV mutation status
Sources: Robbins Pathologic Basis of Disease · Henry's Clinical Diagnosis & Management · Tietz Textbook of Laboratory Medicine · Quick Compendium of Clinical Pathology · Goldman-Cecil Medicine
FMGE exam questions AML ALL CML CLL
Searching the Web
FMGE exam questions AML ALL CML CLL leukemia MCQ previous year
Finding Sources
Finding Sources
Reading Web Page
https://www.prepladder.com/fmge-study-material/previous-year…
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
I now have all the data needed. Here is the complete, high-yield FMGE MCQ bank:
FMGE High-Yield MCQs: AML · ALL · CML · CLL
Exam pattern note: FMGE tests leukemias through clinical vignettes, pathognomonic findings, genetic markers, and drug mechanisms. The questions below are modeled on actual FMGE/NExT previous year patterns.
SECTION A: AML — Acute Myeloid Leukemia
Q1. A 15-year-old boy presents with bleeding gums, subconjunctival hemorrhage, and purpuric rash. CBC shows Hb 6.4 g/dL, TLC 26,500/mm³, platelets 35,000/mm³. Prothrombin time is prolonged. Peripheral smear shows myeloblasts. What is the MOST LIKELY subtype?
- A. Myeloblastic leukemia without maturation (M1)
- B. Myeloblastic leukemia with maturation (M2)
- C. Acute Promyelocytic Leukemia (M3) ✓
- D. Myelomonocytic leukemia (M4)
Explanation: Bleeding + prolonged PT + myeloblasts = DIC → classic for APL (M3). DIC occurs because the abnormal promyelocytes release procoagulant tissue factor. The t(15;17) PML::RARA mutation is the hallmark.
- Henry's Clinical Diagnosis & Management by Laboratory Methods
Q2. Which of the following is PATHOGNOMONIC for AML?
- A. Smudge cells
- B. Philadelphia chromosome
- C. Auer rods ✓
- D. Hyperdiploidy
Explanation: Auer rods are eosinophilic, needle-like cytoplasmic inclusions derived from MPO-positive primary granules. They are pathognomonic for AML and are NEVER seen in ALL (with rare ETP-ALL exception).
- Robbins Pathologic Basis of Disease
Q3. A patient with AML is started on a drug that causes differentiation of malignant promyelocytes into mature neutrophils. The drug is acting on which translocation?
- A. t(9;22)
- B. t(8;21)
- C. inv(16)
- D. t(15;17) ✓
Explanation: ATRA (All-Trans Retinoic Acid) overcomes the differentiation block caused by the PML-RARα fusion protein in APL [t(15;17)], forcing abnormal promyelocytes to mature. This is the basis of the "differentiation therapy" paradigm.
Q4. The MINIMUM blast percentage in bone marrow required to diagnose AML is:
- A. 5%
- B. 10%
- C. 20% ✓
- D. 30%
Explanation: WHO criteria require ≥20% blasts in bone marrow or peripheral blood to diagnose AML. (Exception: if AML-defining genetics like t(15;17), t(8;21), inv(16) are present, diagnosis is made regardless of blast count.)
Q5. In AML, which marker is detected by flow cytometry that confirms MYELOID lineage?
- A. CD19
- B. TdT
- C. CD33 / CD13 / MPO ✓
- D. CD10
Explanation: CD13, CD33, CD117, and MPO are myeloid markers. TdT and CD19/CD10 are lymphoid (B-ALL). This distinction is the foundation of the AML vs. ALL differential.
Q6. A patient with AML has gingival hypertrophy, skin infiltrates, and a very high monocyte count. Which FAB subtype is MOST LIKELY?
- A. M1
- B. M2
- C. M3
- D. M4/M5 (Monocytic/Myelomonocytic) ✓
Explanation: Gingival hypertrophy is characteristic of M4 (myelomonocytic) and M5 (monocytic) AML due to tissue infiltration by monocytes/monoblasts. These cells are NSE (non-specific esterase) positive.
Q7. The BEST PROGNOSIS in AML is associated with which cytogenetic finding?
- A. del(17p) / TP53 mutation
- B. FLT3-ITD mutation
- C. t(15;17) PML::RARA ✓
- D. KMT2A (11q23) rearrangement
Explanation: APL with t(15;17) has a "Very favorable" prognosis because ATRA + arsenic trioxide achieves >90% long-term cure rates. FLT3-ITD and TP53 mutations carry the worst prognosis.
SECTION B: ALL — Acute Lymphoblastic Leukemia
Q8. ALL is MOST COMMON in which age group?
- A. Neonates
- B. Children aged 2-5 years ✓
- C. Young adults 20-30 years
- D. Elderly >60 years
Explanation: ALL is the most common cancer of childhood, peaking at 2-3 years of age. It accounts for ~25% of all childhood malignancies.
- Swanson's Family Medicine Review
Q9. A 4-year-old presents with bone pain, pallor, and lymphadenopathy. Bone marrow biopsy shows sheets of small cells with scant cytoplasm and fine chromatin. MPO is NEGATIVE. TdT is POSITIVE. Diagnosis?
- A. AML
- B. ALL ✓
- C. CML
- D. CLL
Explanation: TdT+ (Terminal deoxynucleotidyl transferase) is the hallmark of ALL lymphoblasts. MPO negativity excludes AML. Scant cytoplasm and fine chromatin with prominent lymphadenopathy in a child strongly favors ALL.
Q10. The BEST PROGNOSIS in childhood ALL is associated with:
- A. t(9;22) Philadelphia chromosome
- B. Hypodiploidy (<46 chromosomes)
- C. Hyperdiploidy (>50 chromosomes) ✓
- D. KMT2A rearrangement
Explanation: Hyperdiploidy (>50 chromosomes) in B-ALL carries the best prognosis in childhood ALL. Hypodiploidy carries the worst prognosis. t(9;22) / Philadelphia chromosome in ALL (25% of adult ALL) carries poor prognosis and requires TKI addition.
Q11. A 16-year-old male presents with anterior mediastinal mass + lymphoblasts in bone marrow. The MOST LIKELY diagnosis is:
- A. Hodgkin's lymphoma
- B. T-cell ALL ✓
- C. B-cell ALL
- D. CML blast crisis
Explanation: T-ALL typically presents in adolescent males with a thymic (anterior mediastinal) mass. T-ALL cells express CD3, CD7, TdT. B-ALL does NOT produce a mediastinal mass.
Q12. The immunophenotype of B-ALL is:
- A. CD3+, CD7+, TdT+
- B. CD19+, CD10+, TdT+, MPO- ✓
- C. CD13+, CD33+, MPO+
- D. CD5+, CD23+, surface Ig dim
Explanation: B-ALL = CD19+ (B-cell), CD10+ (CALLA = Common Acute Lymphoblastic Leukemia Antigen), TdT+, MPO-. CD10 positivity is one of the most tested markers in FMGE. (Option D describes CLL.)
Q13. Which translocation in childhood ALL carries an EXCELLENT prognosis?
- A. t(9;22) BCR::ABL1
- B. KMT2A rearrangement
- C. t(12;21) ETV6::RUNX1 ✓
- D. t(1;19) PBX1::TCF3
Explanation: t(12;21) / ETV6::RUNX1 is the most common translocation in childhood B-ALL (~25%) and carries an excellent prognosis (~90% cure rate). It cannot be detected by standard karyotype - requires FISH or PCR.
Q14. CNS prophylaxis is a critical part of ALL treatment because:
- A. The blood-brain barrier concentrates drugs in the CSF
- B. Leukemic blasts can sanctuary in the CNS and cause relapse ✓
- C. ALL arises in the leptomeninges
- D. Chemotherapy is ineffective in all tissues
Explanation: The CNS is a pharmacological sanctuary - many chemotherapy agents cannot cross the blood-brain barrier effectively, allowing residual blasts to survive and cause CNS relapse. Intrathecal methotrexate is the cornerstone of CNS prophylaxis.
SECTION C: CML — Chronic Myeloid Leukemia
Q15. A 30-year-old male complains of fatigue for 1 year. He has massive splenomegaly. CBC: TLC = 1,50,000/µL with 60% neutrophils, 6% basophils, 4% eosinophils, myeloblasts + myelocytes + metamyelocytes present. The MOST LIKELY diagnosis is:
- A. ALL
- B. AML
- C. CML ✓
- D. CLL
Explanation: This is a classic FMGE/PYQ-confirmed question. The triad of massive splenomegaly + markedly elevated WBC + full myeloid spectrum (myelocyte bulge) + basophilia/eosinophilia in a young adult = CML. Confirmed by Philadelphia chromosome/BCR::ABL1 PCR.
- PrepLadder FMGE PYQ (Actual previous year question)
Q16. "College girl appearance" / "Garden party appearance" of leucocytes on peripheral smear is seen in:
- A. CLL
- B. ALL
- C. AML
- D. CML ✓
Explanation: The peripheral smear in CML shows leukocytes at all stages of maturation - from myeloblasts to mature neutrophils - described as a "garden party / college girl appearance" because all age groups (maturation stages) are present.
- PrepLadder FMGE PYQ (Actual previous year question)
Q17. The Philadelphia chromosome is formed by:
- A. Trisomy of chromosome 22
- B. Deletion of chromosome 9
- C. Reciprocal translocation t(9;22)(q34;q11) ✓
- D. Inversion of chromosome 16
Explanation: The Philadelphia chromosome is the first reproducible chromosomal abnormality identified in human malignancy. The t(9;22) moves the ABL1 gene on chromosome 9 adjacent to BCR on chromosome 22, producing the BCR::ABL1 constitutively active tyrosine kinase (p210).
Q18. The LAP (Leukocyte Alkaline Phosphatase) score in CML is:
- A. Low / decreased ✓
- B. Elevated
- C. Normal
- D. Absent
Explanation: Low LAP score is a hallmark of CML and is the key test distinguishing CML from leukemoid reaction (where LAP is HIGH). In blast crisis of CML, LAP may rise. This is one of the most frequently tested CML facts in FMGE.
Q19. Imatinib (Gleevec) acts by:
- A. Alkylating DNA
- B. Inhibiting topoisomerase II
- C. Competitive inhibition of the ATP-binding site of BCR::ABL tyrosine kinase ✓
- D. Blocking thymidylate synthase
Explanation: Imatinib is the prototype targeted therapy for CML. It competitively inhibits the ATP-binding site of BCR::ABL kinase, preventing phosphorylation of downstream substrates. Also active against PDGFR and c-KIT.
- Katzung's Basic and Clinical Pharmacology
Q20. A CML patient on imatinib develops resistance. Sequencing shows a T315I mutation in BCR::ABL. The drug of choice is:
- A. Dasatinib
- B. Nilotinib
- C. Bosutinib
- D. Ponatinib ✓
Explanation: The T315I "gatekeeper mutation" confers resistance to ALL first- and second-generation TKIs (imatinib, nilotinib, dasatinib, bosutinib). Ponatinib (3rd gen) is the only TKI active against T315I.
Q21. In CML blast crisis, what PERCENTAGE of blasts in the bone marrow is required?
- A. >10%
- B. >15%
- C. >20% ✓
- D. >30%
Explanation: CML blast crisis is defined as ≥20% blasts in blood or bone marrow, OR a tissue infiltrate of blasts (chloroma). At this stage, CML behaves like acute leukemia - 70% AML-type, 30% ALL-type.
Q22. Minimal Residual Disease (MRD) monitoring in CML in remission is done by:
- A. Conventional cytogenetics (karyotype)
- B. FISH for BCR::ABL1
- C. Quantitative RT-PCR for BCR::ABL1 transcript ✓
- D. Flow cytometry for CD34
Explanation: Quantitative RT-PCR is the gold standard for MRD in CML due to its sensitivity (can detect 1 leukemic cell in 10⁵-10⁶ normal cells). A 3-log reduction (major molecular response) at 12 months predicts near-zero progression risk.
SECTION D: CLL — Chronic Lymphocytic Leukemia
Q23. Smudge cells / Basket cells on peripheral smear are MOST CHARACTERISTIC of:
- A. AML
- B. ALL
- C. CML
- D. CLL ✓
Explanation: Smudge (smear/basket) cells are fragile CLL lymphocytes that rupture during smear preparation. They are one of the most pathognomonic peripheral smear findings for CLL.
Q24. The HALLMARK immunophenotype of CLL is:
- A. CD19+, CD10+, CD5-, CD23-
- B. CD19+, CD5+, CD23+, surface Ig dim ✓
- C. CD3+, CD5+, CD23-, TdT+
- D. CD13+, CD33+, CD5-
Explanation: CLL has a unique phenotype: monoclonal B cells (CD19+) that aberrantly co-express the T-cell marker CD5, along with CD23+ and dim surface immunoglobulin. This pattern distinguishes CLL from ALL other B-cell lymphomas.
Q25. CLL is distinguished from Mantle Cell Lymphoma by:
- A. CD5 expression
- B. CD23+ and Cyclin D1- in CLL (vs. CD23- and Cyclin D1+ in MCL) ✓
- C. IgM expression
- D. Bone marrow involvement
Explanation: Both CLL and Mantle Cell Lymphoma (MCL) are CD5+ B-cell neoplasms, making this a classic trap question. Key distinction: CLL = CD23+, Cyclin D1-, LEF1+; MCL = CD23-, Cyclin D1+ [t(11;14)].
Q26. The MOST FAVORABLE cytogenetic finding in CLL is:
- A. del(17p13) [TP53]
- B. del(11q22) [ATM]
- C. del(13q14) ✓
- D. Trisomy 12
Explanation: del(13q14.3) is present in 50% of CLL cases and is associated with the best prognosis when it is the sole abnormality (median survival >10 years). del(17p) with TP53 loss carries the worst prognosis and is resistant to standard chemoimmunotherapy.
Q27. Which CLL molecular marker is associated with POOR prognosis?
- A. Mutated IGHV
- B. Unmutated IGHV + ZAP-70 expression ✓
- C. del(13q14) as sole abnormality
- D. Trisomy 12 alone
Explanation: Unmutated IGHV (naïve B-cell origin) correlates with ZAP-70 expression and an aggressive clinical course. Mutated IGHV (memory B-cell origin) carries a benign, indolent course with longer survival.
Q28. Transformation of CLL into an aggressive large B-cell lymphoma is called:
- A. Reed-Sternberg transformation
- B. Richter's syndrome / Richter transformation ✓
- C. Sézary transformation
- D. Waldenstrom transformation
Explanation: Richter's syndrome is the transformation of CLL/SLL into diffuse large B-cell lymphoma (DLBCL) in ~3-10% of cases. It carries a very poor prognosis (median survival <1 year). Clinically: sudden rapid lymph node enlargement, fever, weight loss, elevated LDH in a CLL patient.
- Robbins Pathologic Basis of Disease
Q29. Autoimmune hemolytic anemia (AIHA) in CLL is mediated by:
- A. NK cell activation
- B. Warm IgG autoantibodies against RBCs (Coombs positive) ✓
- C. Cold IgM agglutinins
- D. Direct blast infiltration of red cells
Explanation: CLL disrupts normal immune function. AIHA occurs in ~10-25% of cases, typically warm-type (IgG), giving a positive Direct Coombs test. This is a treatable complication - steroids or rituximab are used.
SECTION E: COMPARISON / MIXED MCQs (Most Tested in FMGE)
Q30. A child presents with bone pain, pallor, hepatosplenomegaly, and lymphadenopathy. WBC = 80,000/µL with 90% lymphoblasts. MPO is negative. Diagnosis?
- A. CML
- B. CLL
- C. AML
- D. ALL ✓
Explanation: Children + lymphoblasts + MPO negative + hepatosplenomegaly/lymphadenopathy = classic ALL presentation. The markedly elevated WBC with 90% blasts and lymphoid phenotype makes AML (MPO+) and CML (full myeloid spectrum, not blasts) unlikely.
Q31. Which leukemia has the LOWEST LAP score?
- A. AML
- B. ALL
- C. CML ✓
- D. CLL
Explanation: Low LAP (Leukocyte Alkaline Phosphatase) score is a classic hallmark of CML. This is the KEY test to differentiate CML from a leukemoid reaction (high LAP). In ALL and CLL, LAP is normal or variable.
Q32. Auer rods are found in which condition?
- A. ALL
- B. CML
- C. CLL
- D. AML ✓
Explanation: Auer rods = AML, always. They are eosinophilic, needle-like cytoplasmic inclusions derived from fused primary granules (MPO-positive). They are pathognomonic - their presence alone identifies myeloid lineage and excludes ALL.
Q33. Philadelphia chromosome (t(9;22)) is found in which TWO leukemias?
- A. AML and ALL
- B. CLL and CML
- C. CML (defining) and ALL (25% adult cases) ✓
- D. AML and CML
Explanation: Philadelphia chromosome is DEFINING in CML (present in ~95%). It also occurs in 25% of adult ALL (Ph+ ALL), which requires addition of a TKI to chemotherapy. Ph is rare in AML (<1%) and not seen in CLL. This is a classic FMGE trap.
Q34. Which leukemia is most common in:
| Condition | Leukemia |
|---|---|
| Children | ALL |
| Adults (>60 years) | AML |
| Elderly with incidental lymphocytosis | CLL |
| Young adult with massive splenomegaly | CML |
Explanation: Age distribution is one of the MOST TESTED aspects of leukemia in FMGE. Memorize this table.
Q35. Match the peripheral smear finding to the leukemia:
| Smear Finding | Leukemia |
|---|---|
| Auer rods in blast cells | AML |
| Smudge / basket cells | CLL |
| All stages of myeloid maturation (myelocyte bulge) + basophilia | CML |
| Lymphoblasts, scant cytoplasm, TdT+ | ALL |
Q36. Drug of choice for CML chronic phase:
- A. Cytarabine
- B. ATRA
- C. Imatinib (BCR-ABL TKI) ✓
- D. Fludarabine
Q37. Drug of choice for APL (AML-M3):
- A. Imatinib
- B. Cytarabine alone
- C. ATRA + Arsenic Trioxide ✓
- D. Ibrutinib
Explanation: ATRA + Arsenic trioxide (ATO) is now the standard of care for low-risk APL, achieving >90% cure rates without cytotoxic chemotherapy. ATRA induces differentiation; ATO induces differentiation AND apoptosis of APL cells.
Q38. Which leukemia is associated with MASSIVE splenomegaly as a presenting feature?
- A. ALL
- B. AML
- C. CML ✓
- D. CLL
Q39. A 65-year-old man is found incidentally to have WBC = 70,000/µL with 85% mature-appearing small lymphocytes. He feels well. Peripheral smear shows smudge cells. The MOST appropriate next investigation is:
- A. Bone marrow biopsy immediately
- B. Start ibrutinib
- C. Flow cytometry (immunophenotyping) ✓
- D. PCR for BCR::ABL1
Explanation: This is classic CLL. The gold standard for diagnosis and confirmation is flow cytometry showing CD5+/CD19+/CD23+/dim surface Ig B-cells. Treatment is NOT started immediately in early-stage asymptomatic CLL (watch-and-wait policy).
Q40. The Rai staging of CLL - which stage carries the WORST prognosis?
- A. Stage 0 (lymphocytosis only)
- B. Stage I (+ lymphadenopathy)
- C. Stage II (+ splenomegaly)
- D. Stage IV (+ thrombocytopenia) ✓
Explanation: Rai Stage IV = lymphocytosis + thrombocytopenia (or Stage III = anemia). These advanced stages indicate bone marrow failure and carry the worst prognosis. Stage 0 (lymphocytosis alone) = best prognosis, watch-and-wait.
RAPID-FIRE FMGE RECALL TABLE
| High-Yield Point | Answer |
|---|---|
| Most common acute leukemia in children | ALL |
| Most common acute leukemia in adults | AML |
| Most common chronic leukemia in US/Europe | CLL |
| Pathognomonic finding of AML | Auer rods |
| Pathognomonic finding of CLL on smear | Smudge cells |
| Defining marker of CML | Philadelphia chromosome t(9;22) |
| Low LAP score | CML |
| High LAP score | Leukemoid reaction |
| CD10 (CALLA) + leukemia | B-ALL |
| TdT+ leukemia | ALL |
| CD5+ B-cell leukemia | CLL |
| AML associated with DIC | APL (M3) t(15;17) |
| Treatment of APL | ATRA + Arsenic trioxide |
| Treatment of CML | Imatinib |
| T315I mutation → resistant to imatinib | Use Ponatinib |
| CML transforms to acute leukemia | Blast crisis (>20% blasts) |
| CLL transforms to DLBCL | Richter's syndrome |
| Anterior mediastinal mass + leukemia | T-ALL |
| Garden party / College girl smear | CML |
| Worst prognosis AML | TP53 mutation / complex karyotype |
| Best prognosis AML | APL t(15;17) |
| Best prognosis ALL | Hyperdiploidy (>50 chr) |
| Worst prognosis ALL | Hypodiploidy; Ph+ ALL |
| Best prognosis CLL (cytogenetics) | del(13q14) |
| Worst prognosis CLL (cytogenetics) | del(17p13) TP53 |
| Gingival hypertrophy in leukemia | AML M4/M5 (monocytic) |
| Testicular involvement in leukemia | ALL |
Sources: PrepLadder FMGE PYQs · Robbins Pathologic Basis of Disease · Henry's Clinical Diagnosis & Management · Katzung's Basic and Clinical Pharmacology · Goldman-Cecil Medicine · Quick Compendium of Clinical Pathology
This is a shared conversation. Sign in to Orris to start your own chat.