Pheochromocytoma - basic to clinical
Reading File
Finding Sources
Searching PubMed
pheochromocytoma diagnosis management
Searching the Web
pheochromocytoma 2024 2025 guidelines diagnosis treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Loading Image
I now have rich content from multiple authoritative sources. Let me compile the comprehensive answer.
Pheochromocytoma: Basic to Clinical
1. Definition and Etymology
Pheochromocytoma (from Greek phaios = dusky, chroma = color, kytos = cell) is a neoplasm of chromaffin cells of the adrenal medulla that synthesizes and releases catecholamines. The name derives from the characteristic dark-brown discoloration when cut tumor tissue is exposed to potassium dichromate solution (chromaffin reaction). When identical tumors arise from extra-adrenal chromaffin tissue (sympathetic or parasympathetic ganglia), they are called paragangliomas (PGLs). Together they are termed PPGLs - pheochromocytomas and paragangliomas.
- Incidence: 2-8 cases per million per year; found in ~0.2-0.6% of hypertensive patients
- Peak age (sporadic): 40-50 years; familial cases manifest earlier
- Males and females affected equally
"Pheochromocytoma has been described as a biologic time bomb because of the potentially lethal cardiovascular effects of the bioactive compounds secreted by these tumors." - Sabiston Textbook of Surgery
2. The "10% Rule" - Historical and Modern Revisions
Historically, pheochromocytomas were called the "10% tumor":
| Feature | Old "10% Rule" | Modern Reality |
|---|---|---|
| Extra-adrenal | 10% | ~10-15% (up to 20% in familial) |
| Bilateral | 10% | Up to 50% in familial syndromes |
| Malignant | 10% | >10% at diagnosis; up to 20% in extra-adrenal sites |
| Familial | 10% | Now thought to be 30-40% |
| In children | 10% | ~10% |
The 10% rule has been largely revised, especially regarding malignancy and familial rates. Modern genetics have shown far more hereditary cases than previously appreciated.
3. Embryology and Cell of Origin
Pheochromocytomas arise from chromaffin cells (a specialized form of neuroendocrine cell derived from the neural crest). During development, neural crest cells migrate to:
- The adrenal medulla (most common site - ~80-85% of PPGLs)
- Extra-adrenal sympathetic ganglia chain along the aorta
- The organ of Zuckerkandl - the largest collection of extra-adrenal chromaffin tissue, located at the bifurcation of the aorta or the origin of the inferior mesenteric artery (most common site for extra-adrenal paraganglioma)
- Carotid body, jugular-tympanic region, urinary bladder
Normal adrenal medullary chromaffin cells synthesize and store catecholamines in electron-dense secretory granules - the neoplastic counterparts retain this function, often in an exaggerated, dysregulated manner.
4. Genetics and Molecular Pathogenesis
This is now one of the best-characterized heritable tumor syndromes. At least 30-40% of PPGLs harbor germline mutations. Key driver genes fall into two major oncogenic clusters:
Cluster 1 - Pseudohypoxia Pathway (HIF activation)
| Gene | Syndrome | Key Features |
|---|---|---|
| VHL | Von Hippel-Lindau disease | Bilateral PCC, clear cell RCC, hemangioblastomas, pancreatic NETs. ~20% of VHL type 2 develop PPGL |
| SDHB | Hereditary PGL syndrome type 4 | High risk of malignancy (up to 50%); chest/abdomen/pelvis PGL |
| SDHD | Hereditary PGL syndrome type 1 | Head and neck PGL predominance; multiple PGL; imprinting - usually paternal inheritance |
| SDHC | Hereditary PGL syndrome type 3 | Head and neck PGL |
| SDHA | Less common; gastric GIST risk | |
| SDHAF2 | Head/neck PGL; paternal imprinting | |
| EPAS1 (HIF2α) | Sporadic/somatic | Polycythemia-paraganglioma-somatostatinoma triad |
| FH | Fumarate hydratase deficiency | Aggressive, malignant potential |
Cluster 2 - Kinase Signaling / RAS-MAPK-PI3K
| Gene | Syndrome | Key Features |
|---|---|---|
| RET | MEN2A and MEN2B | Bilateral PCC; MTC, PHPT (MEN2A); MEN2B: marfanoid habitus, mucosal neuromas, Hirschsprung |
| NF1 | Neurofibromatosis type 1 | ~2% develop adrenal PCC; café-au-lait spots, Lisch nodules, neurofibromas |
| TMEM127 | Hereditary bilateral PCC | Bilateral adrenal PCC |
| MAX | Bilateral PCC; preferential paternal transmission |
Mechanism summary:
- RET, NF1: enhance growth factor receptor/RAS signaling
- VHL, SDHx, EPAS1: increase HIF activity → pseudohypoxic response → tumor growth
- All pathways ultimately drive uncontrolled chromaffin cell proliferation
Genetic testing is indicated in: bilateral/multiple tumors, extra-adrenal location, age <40 years, family history, or malignant disease. Cascade testing for SDHx, VHL, and RET should be pursued.
5. Pathology
Gross Morphology
- Range from small circumscribed nodules to large hemorrhagic masses weighing several kilograms
- Cut surface: yellow-tan (small tumors) to hemorrhagic, necrotic, cystic (large tumors)
- Fresh tissue + potassium dichromate → dark brown (positive chromaffin reaction)
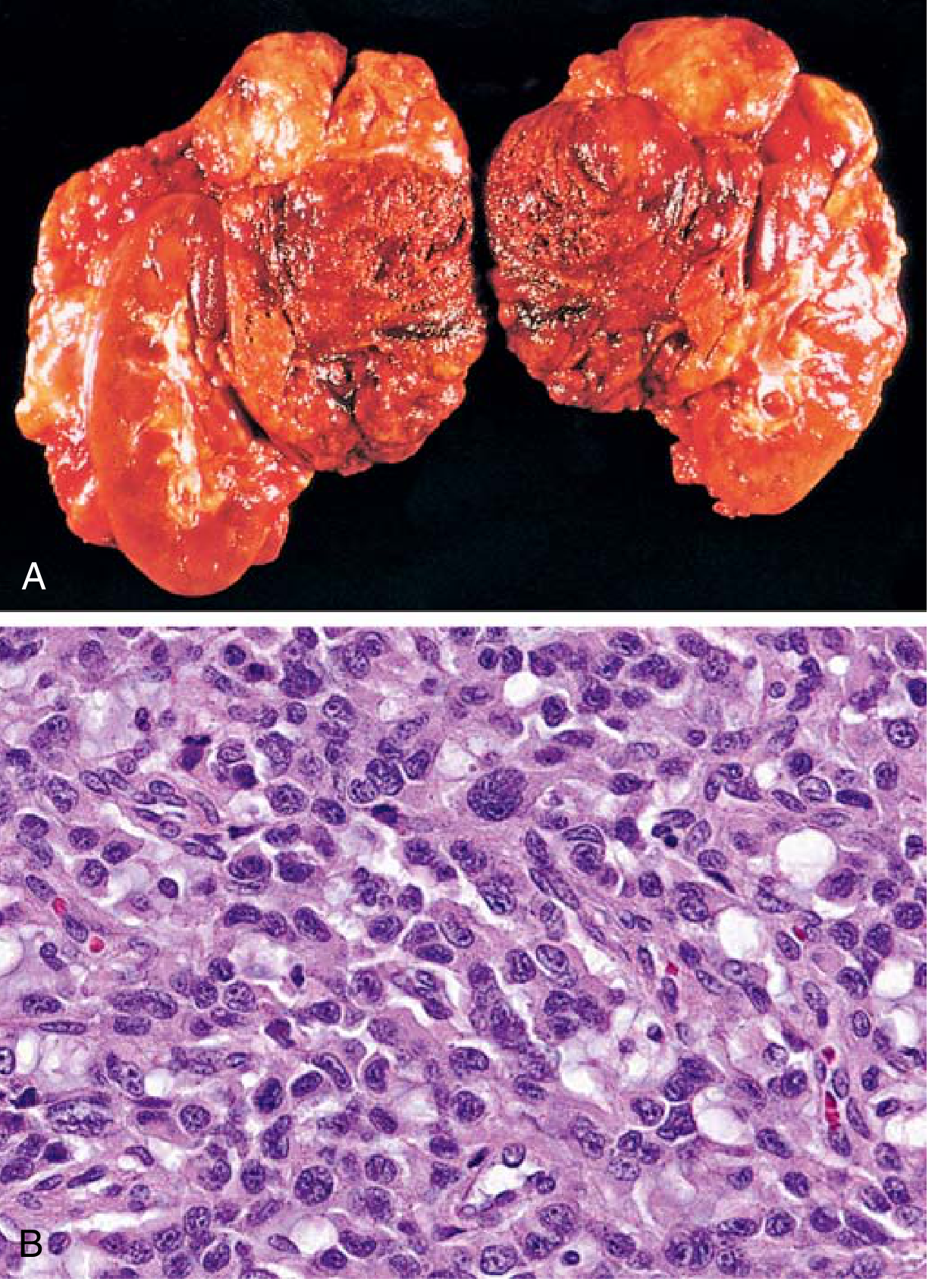
Microscopic Features
- Polygonal to spindle-shaped chromaffin cells with supporting (sustentacular) cells
- Arranged in characteristic "Zellballen" (cell balls) - small nests compartmentalized by a rich vascular network
- Finely granular cytoplasm containing catecholamine-laden granules (highlighted by silver stains; electron-dense on EM)
- Nuclei often pleomorphic - this does NOT indicate malignancy
- Capsular and vascular invasion may be present in benign lesions
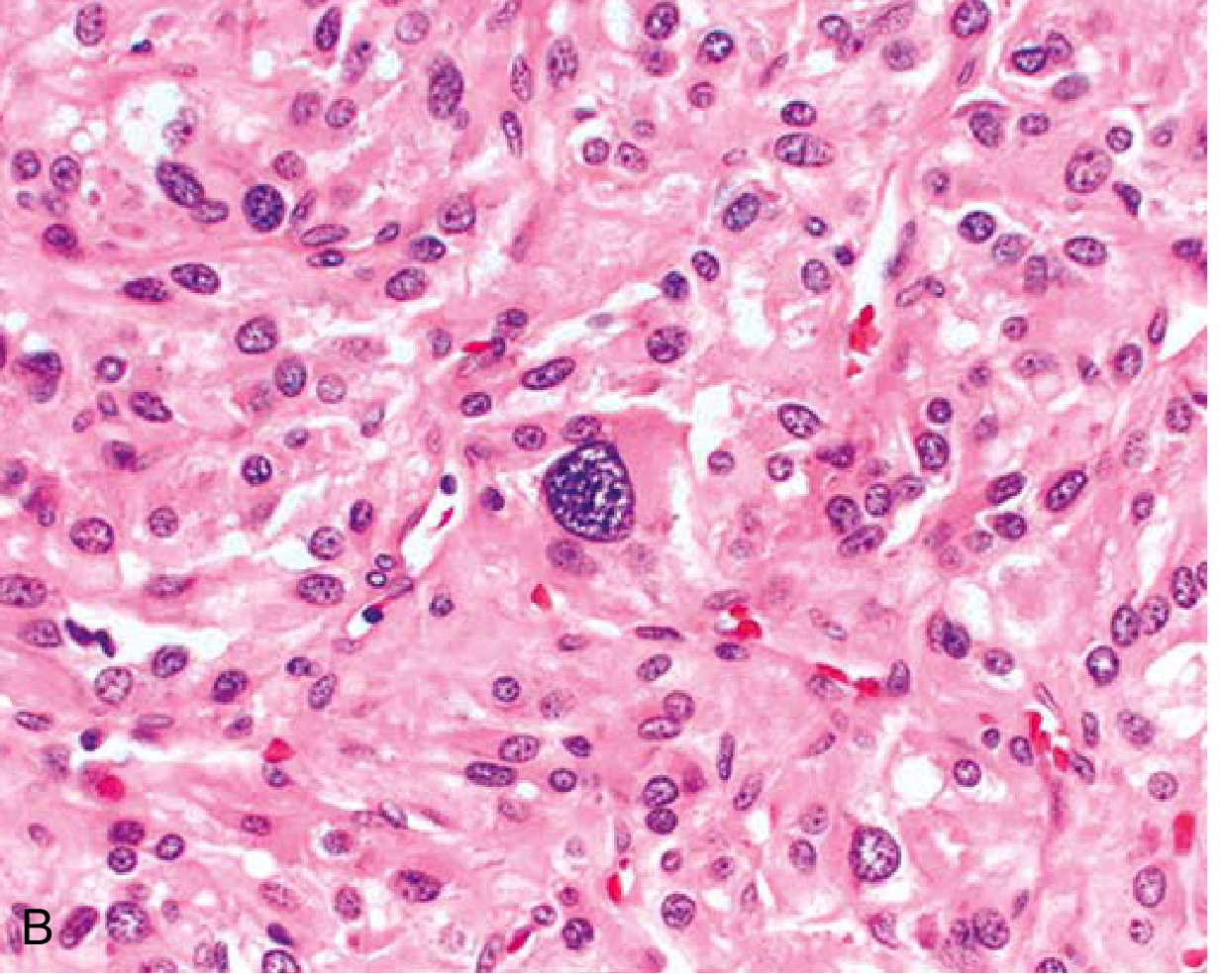
Defining Malignancy
Histology cannot reliably predict malignancy. The ONLY reliable criterion is the presence of metastases (regional lymph nodes, liver, lung, bone) - sites where chromaffin tissue is not normally found. The PASS score (Pheochromocytoma of the Adrenal gland Scaled Score) and GAPP score attempt risk stratification but have limitations.
6. Catecholamine Biochemistry
Understanding catecholamine metabolism is key to both pathophysiology and biochemical diagnosis:
Tyrosine → DOPA → Dopamine → Norepinephrine → Epinephrine
↓ ↓
Normetanephrine Metanephrine
↓ ↓
Vanillylmandelic acid (VMA)
- Rate-limiting step: Tyrosine hydroxylase (converts tyrosine → DOPA)
- Catecholamines are stored in chromaffin granules and released by exocytosis
- Metanephrines (normetanephrine, metanephrine) are formed by continuous O-methylation within the tumor cells even between episodes - this is why they are more sensitive markers than catecholamines themselves
- Final common metabolite: VMA (vanillylmandelic acid), excreted in urine
7. Clinical Features
The Classic Triad (present in ~95% in large series)
- Episodic headache (severe, pounding)
- Diaphoresis (generalized sweating)
- Palpitations / tachycardia
Hypertension Patterns
- Sustained hypertension: most common (~50%)
- Paroxysmal hypertension (episodic "spells"): ~2/3 of patients also have paroxysmal episodes superimposed on chronic hypertension
- ~10% are normotensive (incidentally discovered "silent pheos")
Paroxysmal Episodes ("Spells")
Triggered by: tumor manipulation, exercise, micturition (bladder PGL), Valsalva, certain foods (tyramine), medications (tricyclics, metoclopramide, glucagon, beta-blockers used alone)
- Abrupt BP elevation
- Headache, sweating, palpitations
- Tremor, anxiety, sense of doom
- Abdominal/chest pain, nausea, vomiting
- Duration: minutes to hours
Life-threatening Presentations
- Hypertensive crisis → stroke, ICH
- Acute MI, ventricular fibrillation
- Catecholamine-induced cardiomyopathy (dilated or Takotsubo-like)
- Pulmonary edema
- Pheochromocytoma crisis: multiorgan failure with fever, encephalopathy, shock
Special Presentations
- Micturition syncope/headache: bladder paraganglioma
- Cushing features: ACTH-secreting tumor
- Hypoglycemia: insulin or IGF-secreting component
- Heart failure as presenting complaint (catecholamine cardiomyopathy)
- Incidental adrenal mass on imaging ("incidentaloma")
Physical Exam Clues to Familial Syndromes
| Finding | Syndrome |
|---|---|
| Café-au-lait spots, axillary freckling, Lisch nodules, neurofibromas | NF1 |
| Retinal hemangiomas, cerebellar signs | VHL |
| Mucosal neuromas, marfanoid habitus, thick corneal nerves | MEN2B |
| Port wine stains, subungual fibromas, ash leaf spots, adenoma sebaceum | Tuberous sclerosis |
8. Biochemical Diagnosis
The cornerstone of diagnosis is demonstrating elevated catecholamines or their metabolites.
First-Line Tests (either or both recommended)
| Test | Sensitivity | Specificity | Notes |
|---|---|---|---|
| Plasma free metanephrines | ~99% | ~85-89% | Best for ruling out (high sensitivity); 24h collection not needed; position-dependent - collect supine |
| 24h urine fractionated metanephrines | ~97% | ~91% | Useful as first or confirmatory test; avoids position-related false positives |
"Plasma free metanephrine testing carries an extremely high sensitivity, approaching 99%... the primary usefulness of plasma free metanephrine testing is to exclude pheochromocytoma when the test result is negative." - Sabiston Textbook of Surgery
- Plasma/urine free metanephrine concentrations above 2x the upper limit of normal are diagnostic and essentially confirmatory.
- For borderline results: clonidine suppression test (clonidine 0.3 mg PO → normetanephrine suppresses in essential hypertension but NOT in PPGL)
Second-Line / Supplementary Tests
- 24h urine catecholamines (norepinephrine, epinephrine, dopamine)
- 24h urine VMA (less sensitive than metanephrines)
- Urine dopamine elevated in SDHB-related or dopamine-secreting tumors
Interfering Substances (cause false positives)
- Tricyclic antidepressants, SNRIs
- Labetalol (interferes with HPLC-based assays)
- Levodopa
- Acetaminophen (some assays)
- Physical stress, catecholamine infusions
9. Imaging and Localization
Once biochemically confirmed, imaging is performed to localize the tumor.
Anatomic Imaging
CT scan (preferred first line)
- Sensitivity: 90-100% for adrenal PPGL
- HU >10 on unenhanced CT (pheos are lipid-poor; adenomas typically <10 HU)
- Vigorous early enhancement with <60% venous phase washout
- Typical HU 40-50 on non-contrast CT
- CT neck/chest/abdomen/pelvis (with and without contrast) for complete survey
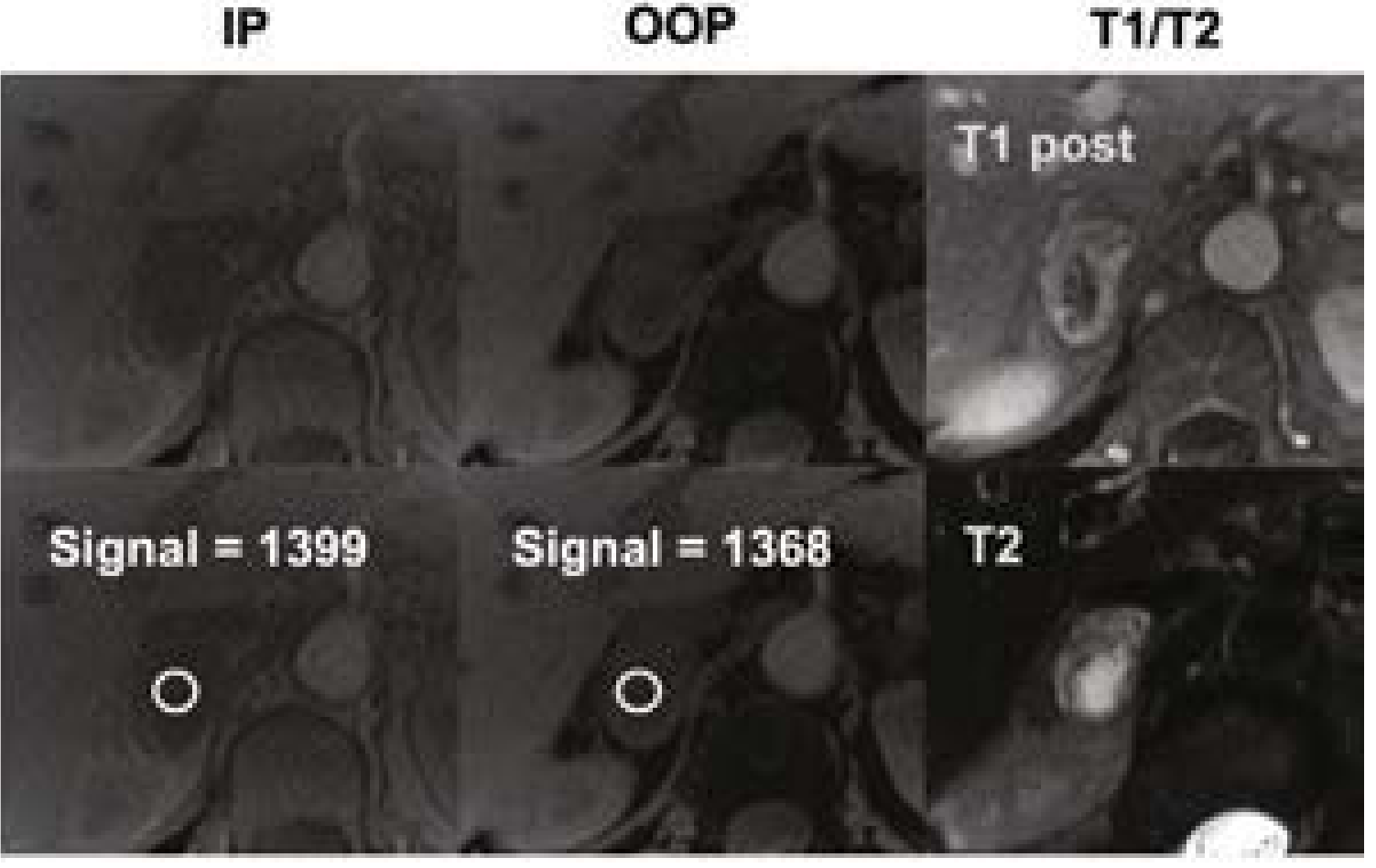
MRI
- Preferred in: pregnant or lactating patients, pediatric patients, CT contrast allergy, desire to avoid radiation
- Classic: very bright on T2-weighted sequences ("light bulb sign") due to high vascularity and water content - but not always present
- No chemical shift dropout (unlike adrenal adenoma)
- Higher sensitivity for extra-adrenal PGL than CT
Functional Imaging (when anatomic imaging inconclusive, or for metastatic workup)
| Modality | Best For | Notes |
|---|---|---|
| ¹²³I-MIBG scintigraphy | SDHB-negative sporadic PCC; pre-treatment planning for MIBG therapy | ~90% sensitivity for adrenal; lower for SDHB-mutated |
| ¹⁸F-FDG PET/CT | Metastatic/malignant PPGL; SDHB-mutated tumors | Best for SDHB-related disease |
| ¹⁸F-DOPA PET/CT | Sporadic, non-metastatic | High sensitivity for head/neck PGL |
| ⁶⁸Ga-DOTATATE/DOTATOC PET/CT | All PPGLs; best overall sensitivity | Recommended when other modalities negative or for metastatic staging |
10. Preoperative Management
This is the most clinically critical step - inadequate preparation is a major cause of perioperative mortality.
Step 1: Alpha-Adrenergic Blockade (MANDATORY - start FIRST)
Phenoxybenzamine (preferred at many centers)
- Non-selective, non-competitive, irreversible alpha-blocker
- Start 10 mg BID, titrate every 2-3 days up to 40 mg TID
- Long half-life (24h) allows decay in parallel with catecholamines postoperatively
- Side effects: postural hypotension, nasal congestion, reflex tachycardia
- Duration: at least 10-14 days preoperatively
Selective alpha-1 blockers (doxazosin, prazosin)
- Increasingly preferred due to cost/availability issues with phenoxybenzamine
- Selective alpha-1 blockade; less reflex tachycardia
- More intraoperative hemodynamic fluctuation; requires excellent anesthesia-surgery communication
Calcium channel blockers (amlodipine, nicardipine) may be added for residual BP control.
Step 2: Beta-Blockade (ONLY AFTER alpha-blockade established)
- For persistent tachycardia (often in epinephrine-predominant tumors)
- NEVER give beta-blocker first - unopposed alpha-adrenergic tone will worsen hypertension and may precipitate crisis
Volume Expansion
- Alpha-blockade creates venous capacitance increase - encourage high-salt diet and oral fluid intake
- IV fluids preoperatively if significant orthostasis
Goals of Preoperative Optimization
- BP <130/80 mmHg seated, no orthostatic drop >10 mmHg
- HR 60-70 bpm seated; no more than 80 standing
- No ST/T wave changes on ECG
- Nasal stuffiness = sign of adequate alpha-blockade
11. Surgical Management
Surgery is curative in >90% of pheochromocytoma cases.
Approach
- Laparoscopic adrenalectomy is now the standard of care for most adrenal pheochromocytomas (tumors <6 cm, no local invasion)
- Open adrenalectomy for large (>6 cm), invasive, or malignant-appearing tumors, or those requiring en-bloc resection
- Cortical-sparing adrenalectomy is recommended for bilateral tumors (to preserve adrenocortical function) - supported by recent meta-analysis [PMID 40214691, 2025]
- For extra-adrenal PGL: laparoscopic or open resection depending on location
Intraoperative Considerations
- Arterial line, large-bore IV access, central line
- Minimize tumor manipulation
- Intraoperative hypertension: IV phentolamine (non-selective alpha), sodium nitroprusside, or nicardipine
- Intraoperative tachyarrhythmias: esmolol, lidocaine
- Post-ligation hypotension (after adrenal vein ligation): sudden withdrawal of catecholamines → vasodilation + increased venous capacitance → cardiovascular collapse. Manage with aggressive IV fluids and vasopressors (vasopressin often more effective due to preoperative alpha-blockade)
Postoperative Care
- Hypoglycemia monitoring (catecholamine-mediated gluconeogenesis/glycogenolysis resolves)
- Hypotension: treat with IV fluids, vasopressors
- Watch for adrenal insufficiency after bilateral adrenalectomy
12. Malignant Pheochromocytoma
- Diagnosed only by presence of metastases - not histology
- Metastatic sites: liver, bone, lung, lymph nodes
- SDHB mutation = strongest predictor of malignant behavior (up to 50% metastatic rate)
- 5-year survival for metastatic PPGL: ~35-60%
Treatment of Metastatic Disease
| Modality | Notes |
|---|---|
| ¹³¹I-MIBG therapy | For MIBG-avid tumors; 131I-iobenguane (Azedra) FDA-approved; partial/complete responses ~25% |
| ⁶⁸Ga-DOTATATE-guided ¹⁷⁷Lu-DOTATATE (PRRT) | Emerging for somatostatin receptor-positive tumors |
| CVD chemotherapy (Cyclophosphamide + Vincristine + Dacarbazine) | Standard cytotoxic regimen; ~57% biochemical response, ~35% partial radiographic response |
| Sunitinib | TKI with activity; SDHB-mutated more likely to respond |
| Metyrosine (alpha-methyl-p-tyrosine) | Inhibits tyrosine hydroxylase → reduces catecholamine synthesis; useful for symptomatic control |
| Bone-directed therapy | Bisphosphonates, denosumab for bone metastases |
13. Special Populations
Pheochromocytoma in Pregnancy
- Rare but potentially fatal - 12% fetal mortality if diagnosed antenatally
- Unrecognized PPGL in pregnancy: 27x increase in maternal and fetal complications
- Management: alpha-blockade is the cornerstone
- If diagnosed <24 weeks: laparoscopic adrenalectomy in 2nd trimester is an option
- If diagnosed in 3rd trimester: alpha-blockade until cesarean delivery, then tumor resection
Pediatric PPGL
- 70-80% are hereditary (vs ~40% in adults)
- Higher rate of bilateral, multifocal, extra-adrenal, and metastatic disease
- Genetic testing is essentially universal
14. Familial Syndromes Summary
| Syndrome | Gene | Chromosome | PPGL Features | Other Tumors |
|---|---|---|---|---|
| MEN2A | RET | 10q11 | Bilateral adrenal PCC (~50%) | Medullary thyroid cancer, PHPT |
| MEN2B | RET | 10q11 | Bilateral adrenal PCC | MTC, mucosal neuromas, marfanoid habitus, Hirschsprung |
| VHL type 2 | VHL | 3p25 | Bilateral PCC (~20%) | Hemangioblastomas, clear cell RCC, pancreatic NETs |
| NF1 | NF1 | 17q11 | Adrenal PCC (~2%) | Neurofibromas, café-au-lait, CNS gliomas |
| Hereditary PGL 1 | SDHD | 11q23 | Head/neck PGL, multiple PGL | Maternal imprinting |
| Hereditary PGL 3 | SDHC | 1q23 | Head/neck PGL | - |
| Hereditary PGL 4 | SDHB | 1p36 | Abdominopelvic PGL, malignant | GI stromal tumors, RCC |
| SDH5 (PGL2) | SDHAF2 | 11q13 | Head/neck PGL | Paternal imprinting |
15. Postoperative Follow-up
- Biochemical testing (plasma free metanephrines or 24h urine) at 2-6 weeks post-op to confirm biochemical cure
- Annual biochemical surveillance for life (recurrence risk ~17%)
- Imaging if biochemistry becomes elevated again
- Genetic counseling and cascade family screening for all mutation carriers
Key Clinical Pearls
- Never give beta-blockers alone to a hypertensive patient with suspected pheo - it will worsen the crisis by unopposed alpha-stimulation.
- Plasma free metanephrines are the most sensitive test (99%) - a negative result essentially rules out pheo.
- SDHB mutation = think malignancy, pursue whole-body functional imaging.
- The chromaffin reaction (dichromate turns tissue brown) = the basis of the tumor's name.
- Histologic pleomorphism and capsular invasion do not define malignancy - only metastasis does.
- "Silent pheos" (normotensive, incidentally found) are increasing in frequency as cross-sectional imaging becomes more widespread.
- Postoperative hypoglycemia and hypotension are expected consequences of sudden catecholamine withdrawal.
- In bilateral disease, cortical-sparing adrenalectomy is preferred to avoid lifelong steroid replacement.
- Bladder paraganglioma classically presents with micturition-induced hypertensive spells or syncope.
- Any adrenal incidentaloma workup must include pheochromocytoma screening before biopsy or surgery to prevent triggering a crisis.
Sources:
- Robbins & Kumar Basic Pathology (9780323790185), pp. 761-763
- Sabiston Textbook of Surgery (9780443124341), Adrenal Surgery chapter
- Current Surgical Therapy 14e (9780323796835), pp. 869-872
- Brenner and Rector's The Kidney, 2-vol (9780323532655), pp. relevant sections
- Campbell-Walsh-Wein Urology (9780323546423), Chapter 106
Recent evidence (2025):
- A 2025 meta-analysis [PMID 40214691] on partial adrenalectomy for pheochromocytoma confirms cortical-sparing surgery has comparable oncological outcomes to total adrenalectomy with preserved adrenal function - supporting its use in bilateral disease.
- Japan Endocrine Society [PMID 41083371] published updated 2025 guidelines on PPGL diagnosis and management, incorporating ⁶⁸Ga-DOTATATE PET as a major imaging advance and updating genetic testing recommendations.
Investigations of pheochromocytoma
Finding Sources
Finding Sources
Searching PubMed
pheochromocytoma biochemical diagnosis metanephrine imaging
Reading File
Reading File
Searching PubMed
pheochromocytoma diagnosis guidelines 2024
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Excellent - I now have comprehensive data from Sabiston, Campbell-Walsh, Schwartz, Goldman-Cecil, and Current Surgical Therapy including the precise cutoff values table, test characteristics table, and the diagnostic algorithm. Let me compile the comprehensive, focused answer.
Investigations of Pheochromocytoma
Investigations follow a two-phase approach: first biochemical confirmation (always before imaging), then anatomical and functional localization.
Critical Rule: Biochemical diagnosis MUST precede imaging. Never proceed to biopsy of any adrenal mass before excluding pheochromocytoma - catecholamine release during needle biopsy can precipitate a fatal hypertensive crisis.
Phase 1 - Biochemical Diagnosis
Why Metanephrines, Not Catecholamines?
Catecholamines are released episodically from pheochromocytomas - between attacks, plasma catecholamines may be normal. However, O-methylation of catecholamines to metanephrines is a continuous, uninterrupted process within the tumor cells (catalyzed by COMT), independent of secretory bursts. Metanephrines therefore leak continuously into the circulation even when catecholamines are not being actively secreted, making them far more sensitive markers.
- Norepinephrine → Normetanephrine (via COMT)
- Epinephrine → Metanephrine (via COMT)
- Both → VMA (vanillylmandelic acid) after further MAO metabolism
- The term "free" = unconjugated (not sulfonated); "fractionated" = individual subtype measurements (normetanephrine and metanephrine separately, not as a combined total)
Test 1: Plasma Free Metanephrines (First-Line)
The most sensitive single test available.
| Parameter | Value |
|---|---|
| Sensitivity | 99% (sporadic); 97% (hereditary) |
| Specificity | 82-89% (falls to ~77% in patients >60 years) |
| Cutoff - Metanephrine | 0.3 nmol/L (59 µg/L) |
| Cutoff - Normetanephrine | 0.6 nmol/L (112 µg/L) |
| Result interpretation | Positive if either or both values are elevated |
| Diagnostic threshold | Values >2x upper limit of normal are essentially confirmatory |
Key practical points:
- Collect supine after 30 minutes of rest (standing increases normetanephrine by ~2x physiologically)
- Discontinue interfering medications if possible before testing
- A negative result effectively excludes pheochromocytoma - the primary value of this test is its negative predictive power
- False positive rate is high: in a tertiary hypertension clinic, an estimated 97% of patients with abnormal plasma metanephrines do NOT have pheochromocytoma - because the disease is rare (~0.2% of hypertensives)
- False positives outnumber true positives by as much as 30:1 when used as a population screening tool
Use in hereditary/high-risk screening: Plasma metanephrines preferred over urine for patients with known genetic mutations (VHL, SDHx, RET) due to higher sensitivity for small or biochemically subtle tumors.
Test 2: 24-Hour Urine Fractionated Metanephrines + Catecholamines (First-Line / Confirmatory)
The most specific combined approach for confirmation.
| Test | Sensitivity | Specificity |
|---|---|---|
| Urine fractionated metanephrines (hereditary) | 96% | 82% |
| Urine fractionated metanephrines (sporadic) | 97% | 45% |
| Urine catecholamines (hereditary) | 79% | 96% |
| Urine catecholamines (sporadic) | 91% | 75% |
| Urine total metanephrines | 60-88% | 89-97% |
| Urine VMA | 46-77% | 86-99% |
(Data from Lenders et al., JAMA 2002, multicenter cohort n=858)
Cutoff values (set at ~2x the upper 95% reference range):
| Test | Cutoff | Sensitivity | Specificity |
|---|---|---|---|
| 24h urine metanephrines (total) | 1.3 mg/24h | 77% | 93% |
| 24h urine normetanephrine | 1.8 mg/24h | 97% | 91% |
| 24h urine norepinephrine | 170 µg/24h | 86% | 88% |
| 24h urine epinephrine | 35 µg/24h | - | - |
| 24h urine dopamine | 700 µg/24h | - | - |
| 24h urine VMA | 11 mg/24h | 77% | 86% |
A urine collection is considered positive if total metanephrines OR any single catecholamine fraction exceeds its cutoff value.
Practical protocol: Perform two 24-hour urine collections (sensitivity and specificity maintained; ~98% sensitivity, ~98% specificity for the combined 24h urine approach).
Goldman-Cecil example of a positive result (real patient):
Normetanephrine 8760 µg/24h (normal <900 µg), norepinephrine 781 µg/24h (normal <170 µg) - 5-cm right adrenal pheochromocytoma confirmed on CT
Test 3: VMA (Vanillylmandelic Acid) - Urine
- End metabolite of catecholamine degradation (via both MAO and COMT)
- Long used historically but now least sensitive of the catecholamine tests (sensitivity only 46-77%)
- High specificity (86-99%), especially useful when positive
- No longer recommended as a first-line test; largely replaced by metanephrine assays
Test 4: Clonidine Suppression Test (Adjunctive/Confirmatory)
Used when biochemical results are borderline or equivocal after two 24-hour urine collections.
Principle: Clonidine (alpha-2 agonist) suppresses neurogenically mediated catecholamine release (e.g., anxiety, essential hypertension) but does NOT suppress autonomous secretion from a pheochromocytoma.
Protocol:
- Patient supine; baseline plasma normetanephrine measured
- Oral clonidine 0.3 mg administered
- Repeat plasma normetanephrine at 3 hours
Interpretation:
- Normal response (excludes pheo): Normetanephrine falls to <0.61 nmol/L (suppressed)
- Positive (supports pheo): Normetanephrine remains elevated (NOT suppressed) despite clonidine
- A normal clonidine suppression test also defined by total catecholamines falling to <500 pg/mL within 2-3 hours
Test 5: Chromogranin A (Supplementary)
- Acidic monomeric protein co-stored in chromaffin granules and released alongside catecholamines
- Sensitivity ~83%, specificity ~96%
- Useful as a supplementary marker, not first-line
- Elevated in other neuroendocrine tumors (non-specific)
- Can help confirm neuroendocrine origin
Test Hierarchy Summary
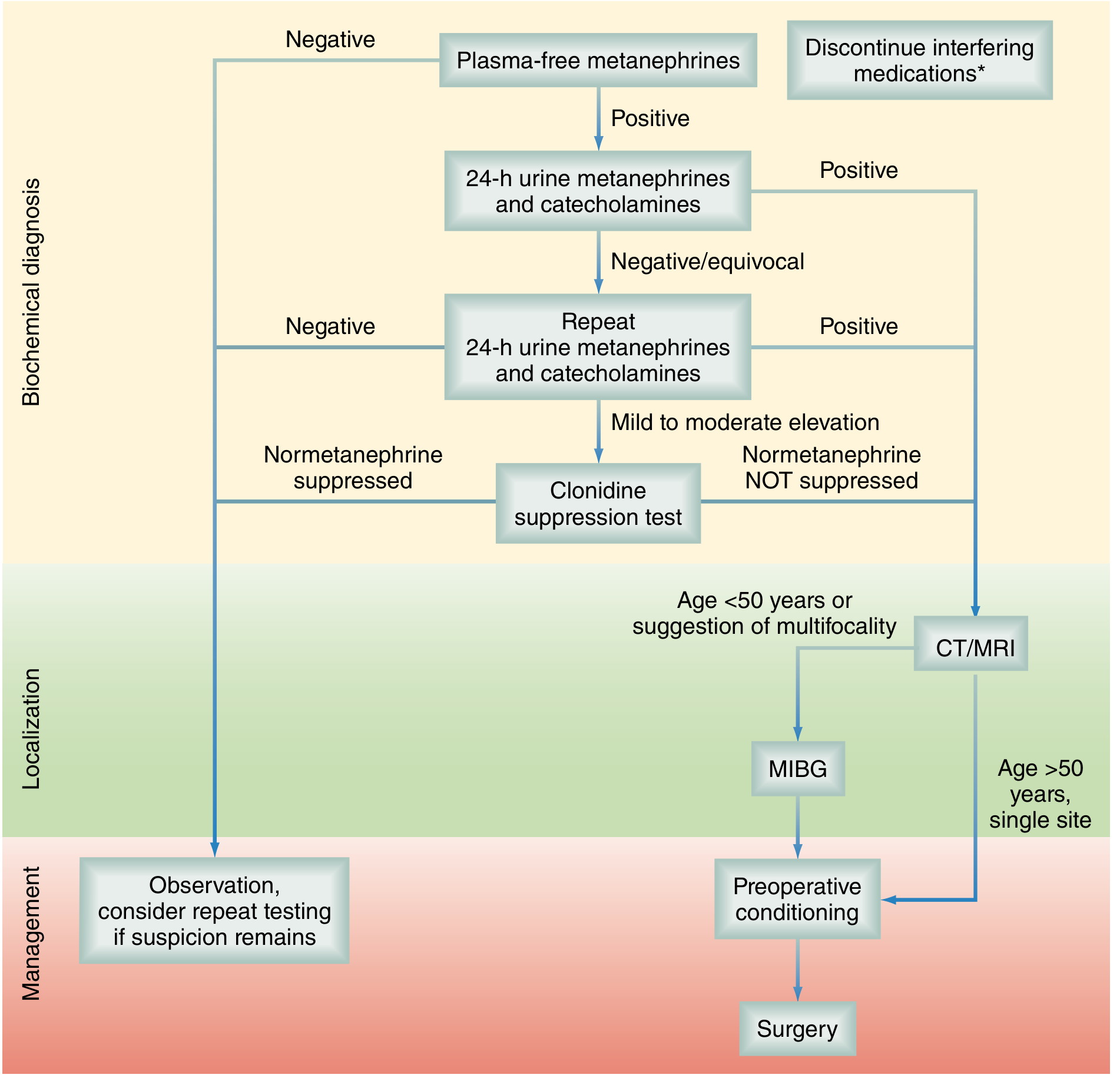
Step 1: Plasma free metanephrines
- If negative → diagnosis excluded (observe; repeat only if suspicion remains high)
- If positive → proceed to Step 2
Step 2: 24-hour urine fractionated metanephrines + catecholamines (perform twice)
- If positive → biochemical diagnosis confirmed → proceed to localization
- If negative/equivocal → proceed to Step 3
Step 3: Clonidine suppression test
- Normetanephrine suppressed → essential hypertension or adrenergic excess (not pheo)
- Normetanephrine NOT suppressed → pheochromocytoma confirmed → proceed to localization
Causes of False-Positive Results
| Category | Examples |
|---|---|
| Medications | Tricyclic antidepressants, SNRIs, phenoxybenzamine, labetalol, levodopa, alpha-methyldopa, cocaine, amphetamines, ephedrine/pseudoephedrine, monoamine oxidase inhibitors |
| Physiological stress | Acute illness, surgery, pain, critical care admission, subarachnoid hemorrhage, MI, preeclampsia, migraine |
| Renal failure | Reduced metanephrine clearance |
| Age >60 years | Specificity of plasma metanephrines falls to ~77% |
| Assay interference | Acetaminophen interferes with some plasma free metanephrine assays |
| Drug withdrawal | Clonidine withdrawal, alcohol withdrawal |
Phase 2 - Anatomical Localization
Imaging is performed only AFTER biochemical confirmation.
CT Scan (First-Line Anatomic Imaging)
Sensitivity: 85-95%, Specificity: 70-100%
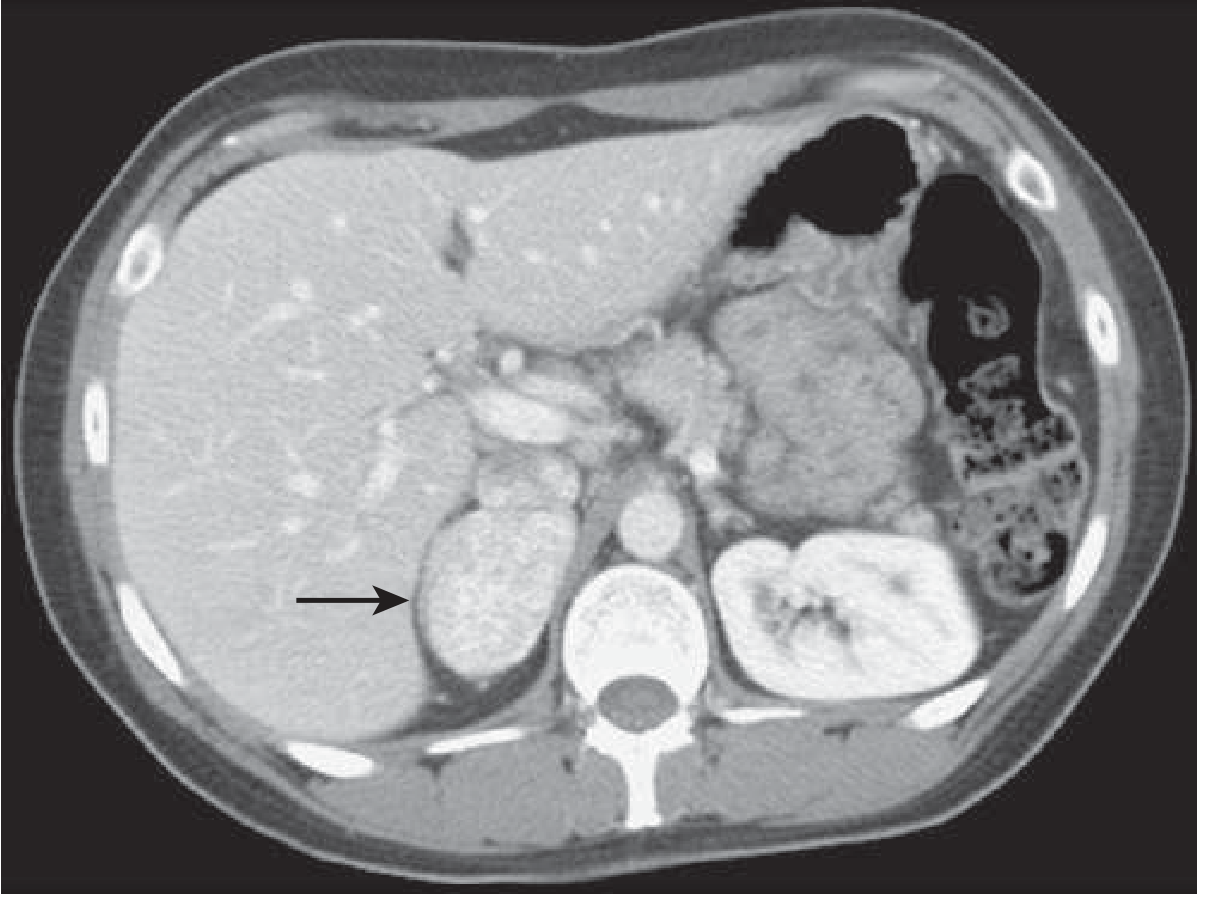
CT characteristics of pheochromocytoma:
- Unenhanced HU: typically 40-50 HU (lipid-poor; adenomas are <10 HU)
- If non-contrast HU >10 → suspicious, warrants further evaluation
- Vigorous early arterial enhancement with post-contrast HU often >100
- Venous phase washout <60% (adenomas wash out >60%)
- Morphology: homogeneous (small tumors) to heterogeneous with necrosis, calcification, cystic change (large tumors)
- Can be solid, cystic, or mixed
CT scanning field: Neck, chest, abdomen, pelvis (with and without contrast) - to include entire sympathetic chain and organ of Zuckerkandl (at aortic bifurcation). Section thickness 5 mm through abdomen/pelvis.
Concern about IV contrast: Older literature advised against IV contrast (fear of precipitating crisis). More recent studies show IV contrast is safe in alpha-blocked patients. Most centers now use contrast CT routinely.
MRI (Preferred in Specific Populations)
Sensitivity: 95-100%, Specificity: nearly 100%
Indications for MRI over CT:
- Pregnant or lactating women (no radiation)
- Pediatric patients
- CT contrast allergy
- Patients declining radiation
- Extra-adrenal paraganglioma (higher MRI sensitivity)
MRI characteristics:
- T2-weighted: classic "light bulb" bright signal due to high vascularity and water content - though not invariably present
- T1: isointense to hypointense relative to liver
- No chemical shift dropout on out-of-phase imaging (no lipid content - key distinction from adrenal adenoma, which loses signal on OOP)
- Post-gadolinium: vigorous enhancement
- Variable: can be homogeneous intermediate, heterogeneous with swirl pattern, or multiple pockets of T2 brightness (cystic)
Comparison: CT vs MRI for Pheo
| Feature | CT | MRI |
|---|---|---|
| Sensitivity | 85-95% | 95-100% |
| Specificity (anatomic only) | 70-100% | ~100% |
| Best for | Operative planning, anatomic definition, initial localization | Pregnancy, children, contrast allergy, extra-adrenal PGL |
| Disadvantage | Radiation, lower sensitivity for extra-adrenal | Less anatomic detail for surgical planning |
Phase 3 - Functional Imaging
Functional imaging is used when:
- CT/MRI negative but biochemistry strongly positive (extra-adrenal location)
- Suspected metastatic or multifocal disease
- High-risk patients: SDHB mutation, large tumor (>5 cm), young age
- Pre-treatment planning for MIBG-directed therapy
¹²³I-MIBG Scintigraphy
- MIBG (metaiodobenzylguanidine) is a structural analogue of guanethidine, taken up by the norepinephrine transporter (NET) and stored in chromaffin granules
- Sensitivity: 77-90% (adrenal); lower for SDHB-mutated and metastatic tumors
- Specificity: 95-100%
- Normal physiological uptake: liver, salivary glands, thyroid, bladder
- MIBG-negative tumors: VHL and SDHB-mutated PPGLs often show "cold" MIBG scans → use ¹⁸F-FDG PET instead
- Also used for pre-treatment staging before ¹³¹I-MIBG therapy
¹⁸F-FDG PET/CT
- Best for SDHB-mutated and metastatic disease
- Aggressive metastatic lesions that lose MIBG uptake often retain FDG avidity
- Sensitivity ~49% overall for sporadic localized pheo, but superior to MIBG for SDHB/malignant
- Particularly useful when MIBG scan is negative
¹⁸F-DOPA (F-dihydroxyphenylalanine) PET/CT
- Highly sensitive for sporadic, non-metastatic adrenal pheochromocytoma
- Sensitivity ~75%; superior to MIBG for head/neck PGL
- Less useful for SDHB-related disease
⁶⁸Ga-DOTATATE/DOTATOC PET/CT (Best Overall)
- Uses radiolabeled somatostatin analogue targeting somatostatin receptors (SSTR2) on neuroendocrine tumors
- Currently the best single functional imaging agent for PPGL localization
- Lesion detection rate: 97.6% - higher than FDG PET (49%) and ¹⁸F-FDOPA (75%)
- Particularly superior for SDHB-associated metastatic disease
- Increasingly available; previously limited to academic centers
- Also guides ¹⁷⁷Lu-DOTATATE PRRT (peptide receptor radionuclide therapy) selection
Functional Imaging Selection Guide
| Clinical Scenario | Best Functional Imaging |
|---|---|
| Sporadic adrenal pheo, single site, no genetic risk | CT/MRI alone sufficient (MIBG optional for >5 cm) |
| Suspected extra-adrenal PGL, CT negative | ⁶⁸Ga-DOTATATE PET/CT or ¹²³I-MIBG |
| SDHB mutation, any PPGL | ¹⁸F-FDG PET/CT + ⁶⁸Ga-DOTATATE |
| VHL mutation | ⁶⁸Ga-DOTATATE PET/CT |
| Pre-¹³¹I-MIBG therapy staging | ¹²³I-MIBG scintigraphy (confirms MIBG avidity) |
| Suspected metastatic disease | ¹⁸F-FDG PET/CT + ⁶⁸Ga-DOTATATE |
| Head and neck PGL | ⁶⁸Ga-DOTATATE PET/CT or ¹⁸F-DOPA |
What NOT to Do
| Pitfall | Consequence |
|---|---|
| Adrenal biopsy before excluding pheo | Catastrophic catecholamine release, hypertensive crisis, death |
| Imaging before biochemical confirmation | Poor specificity of CT/MRI alone - many adrenal incidentalomas are not pheo |
| Using spot urine VMA as a first-line test | Low sensitivity (46-77%); misses many cases |
| Measuring only total catecholamines | Less sensitive than fractionated metanephrines |
| Collecting urine during an acute illness/stress | Physiological catecholamine elevation causes false positives |
| Measuring plasma metanephrines while standing | Normetanephrine is ~2x higher standing than supine |
Additional Investigations (General + Genetic)
Routine Labs
- Electrolytes, renal function (hypokalemia may occur with dopamine excess; renal ischemia from vasoconstriction)
- Blood glucose (catecholamines inhibit insulin; hyperglycemia common)
- CBC (polycythemia in HIF2α/EPAS1 mutations)
- ECG (LVH, ST/T changes, arrhythmias from catecholamine effect)
- Echocardiogram (catecholamine cardiomyopathy evaluation before surgery)
Genetic Testing
Indicated in: bilateral/multiple tumors, extra-adrenal location, age <40 years, family history, malignant PPGL, or any PPGL in a patient with features of a familial syndrome.
Panel: SDHx (SDHB, SDHC, SDHD, SDHA, SDHAF2), VHL, RET, NF1, TMEM127, MAX, HIF2α (EPAS1), FH
Genetic counseling before and after testing is recommended.
Key Numbers to Remember
| Test | Sensitivity | Specificity |
|---|---|---|
| Plasma free metanephrines | 99% | 82-89% |
| 24h urine fractionated metanephrines | 97% | 45-82% |
| 24h urine catecholamines | 91% | 75-96% |
| 24h urine VMA | 77% | 86-99% |
| CT (anatomic) | 85-95% | 70-100% |
| MRI (anatomic) | 95-100% | ~100% |
| ¹²³I-MIBG | 77-90% | 95-100% |
| ⁶⁸Ga-DOTATATE PET | 97.6% | high |
| ¹⁸F-FDG PET | ~49% overall; better for SDHB | - |
Sources:
- Campbell-Walsh-Wein Urology (9780323546423), Table 106.8, pp. 3148-3154
- Sabiston Textbook of Surgery (9780443124341), Table 75.4, pp. 1571-1574
- Schwartz's Principles of Surgery 11e (9781259835346), pp. 1721-1722
- Goldman-Cecil Medicine (9780323930345), Fig. 209-1, pp. 341-355
- Current Surgical Therapy 14e (9780323796835), pp. 869-872
This is a shared conversation. Sign in to Orris to start your own chat.