Pnh
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
paroxysmal nocturnal hemoglobinuria treatment complement inhibitor
Loading Image
Loading Image
I now have everything needed for a comprehensive answer. Here is the full review:
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Definition
PNH is a rare, acquired clonal stem cell disorder in which somatic mutation of the PIGA gene causes deficiency of GPI-anchored complement regulatory proteins on blood cell surfaces, resulting in complement-mediated intravascular hemolysis, thrombosis, and variable cytopenias. It is the only hemolytic anemia caused by an acquired genetic defect.
- Robbins & Kumar Basic Pathology, p. 605
- Goldman-Cecil Medicine, Ch. 146
Pathogenesis
The PIGA Mutation
The PIGA gene on the short arm of the X chromosome encodes an enzyme essential for synthesizing glycosylphosphatidylinositol (GPI), a specialized phospholipid that anchors many proteins to the cell membrane. Because PIGA is X-linked, a single somatic mutation in any hematopoietic stem cell (HSC) is sufficient to eliminate GPI synthesis in all progeny of that clone - red cells, white cells, and platelets alike.
- Nearly 150 different PIGA mutations have been identified
- Most inactivate PIGA completely → PNH III cells (fully deficient)
- Partial deficiency → PNH II cells (3-5x normal complement sensitivity)
- PNH III cells: 15-25x normal complement sensitivity
Why Does the Clone Expand?
Small numbers of PIGA-deficient cells exist in normal individuals (~1 in 50,000 RBCs) but never expand. In PNH patients, a second step - possibly autoimmune attack against GPI-anchored antigens - provides a selective advantage to the mutant clone, allowing it to expand. This explains the frequent association between PNH and aplastic anemia (an autoimmune marrow failure syndrome).
GPI-Linked Proteins Lost in PNH
| Protein | Function |
|---|---|
| CD59 (MIRL, protectin) | Most important - inhibits C3 convertase; blocks C9 polymerization (the final MAC assembly step) |
| CD55 (DAF) | Decay-accelerating factor; breaks down C3/C5 convertases |
| C8-binding protein | Homologous restriction factor |
| CD58, CD14, CD24, CD16a | Additional GPI-linked surface proteins |
| Acetylcholinesterase, LAP | Membrane-associated enzymes |
With CD55 and CD59 absent, the membrane attack complex (C5b-9/MAC) assembles unimpeded on red cell surfaces → intravascular hemolysis.
Clinical Features
Classic Triad
- Hemolytic anemia - chronic intravascular, often with reticulocytosis less than expected
- Venous thrombosis - in unusual sites
- Cytopenias (variable) - neutropenia in 3/5, thrombocytopenia in 2/3 at some point
Hemoglobinuria
The classic "paroxysmal nocturnal" presentation occurs in only ~25% of cases. The rest present with chronic hemolysis without dramatic hemoglobinuria. The nocturnal pattern is attributed to a mild drop in blood pH during sleep (CO2 retention), which activates complement. However, this relationship is not firmly confirmed.
Triggers of hemolysis: infection, surgery, blood transfusion, contrast dye injection, severe exercise, fever, acidosis, hypoxia.
Thrombosis - The Major Killer
- Affects ~40% of patients
- 85% venous, often in unusual sites:
- Hepatic veins (Budd-Chiari syndrome)
- Portal vein
- Cerebral veins
- Abdominal veins
- Mechanism: CD59 absence on platelets → phosphatidylserine externalization → prothrombinase complex assembly; also, free hemoglobin scavenges nitric oxide (NO), causing vasoconstriction and platelet activation
- Abdominal pain in ~1/3 of patients is linked to NO scavenging by free hemoglobin (also causes dysphagia, erectile dysfunction)
Iron Deficiency
Chronic loss of iron in urine as hemosiderinuria → iron deficiency → hypochromic microcytic anemia superimposed on hemolysis. Hemosiderinuria is almost constantly present.
Epidemiology
- Incidence: ~2-5 per million/year
- Any age, most common 10-50 years; mean age at diagnosis ~34 years
- Female:male ratio ~1:1
- Median survival ~20 years after diagnosis
PNH Categories
- Classic PNH - hemolysis predominant, large clone
- PNH in the setting of another bone marrow disorder (aplastic anemia, MDS) - cytopenias predominant; PNH clone usually small
- Subclinical PNH - small GPI-deficient clone, no clinical hemolysis
- Found in 50-60% of aplastic anemia patients
- Found in 15-20% of MDS patients
Diagnosis
Gold Standard: Flow Cytometry (FLAER-based)
The loss of GPI-linked proteins is detected by flow cytometry of peripheral blood. At least two different cell lineages must be evaluated.
Recommended panel:
- Neutrophils: FLAER + CD24 + CD15 + CD45
- Monocytes: FLAER + CD14 + CD64 + CD45
- Red blood cells: CD235a / CD59 (two-color)
FLAER (fluorescent derivative of bacterial proaerolysin) binds the GPI anchor itself directly - the key reagent for high-sensitivity testing.
Flow cytometry findings in PNH:
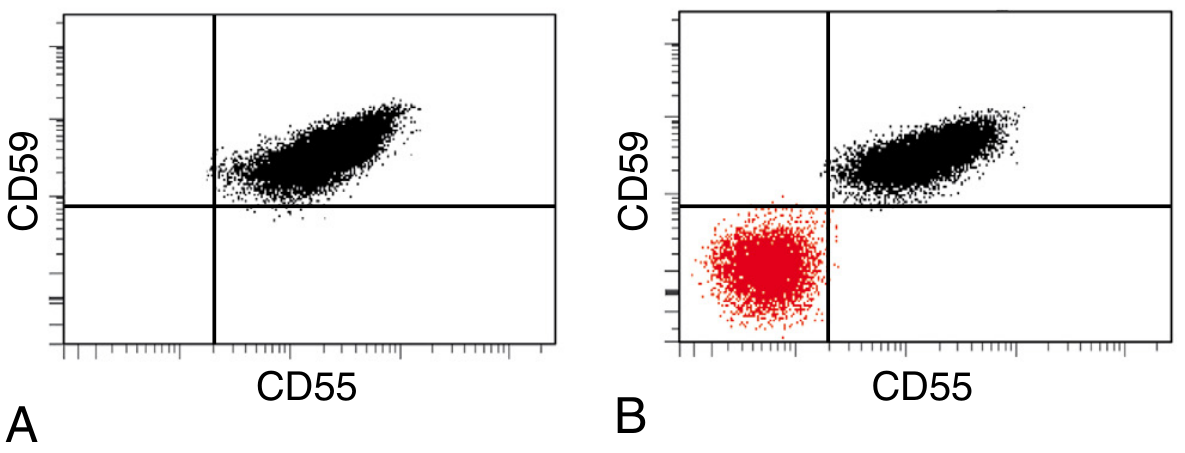
Panel A (normal): All red cells express CD55 and CD59 normally. Panel B (PNH): A large population (red) is negative for both CD55 and CD59 - the PNH clone.
Other Lab Findings
| Finding | Explanation |
|---|---|
| Normocytic anemia (or hypochromic/microcytic) | Hemolysis + iron deficiency |
| Reticulocytosis (less than expected) | Marrow suppression may coexist |
| Hemosiderinuria | Chronic iron loss in urine |
| Negative direct antiglobulin (Coombs) test | Distinguishes PNH from autoimmune hemolysis |
| Elevated LDH | Intravascular hemolysis |
| Low haptoglobin | Free hemoglobin binding |
| Pancytopenia | Common during disease course |
Treatment
Anti-Complement Therapy (Disease-Modifying)
The major advance in PNH management came with eculizumab (approved 2007) - a humanized monoclonal antibody that blocks C5, preventing MAC formation.
Mechanism comparison:
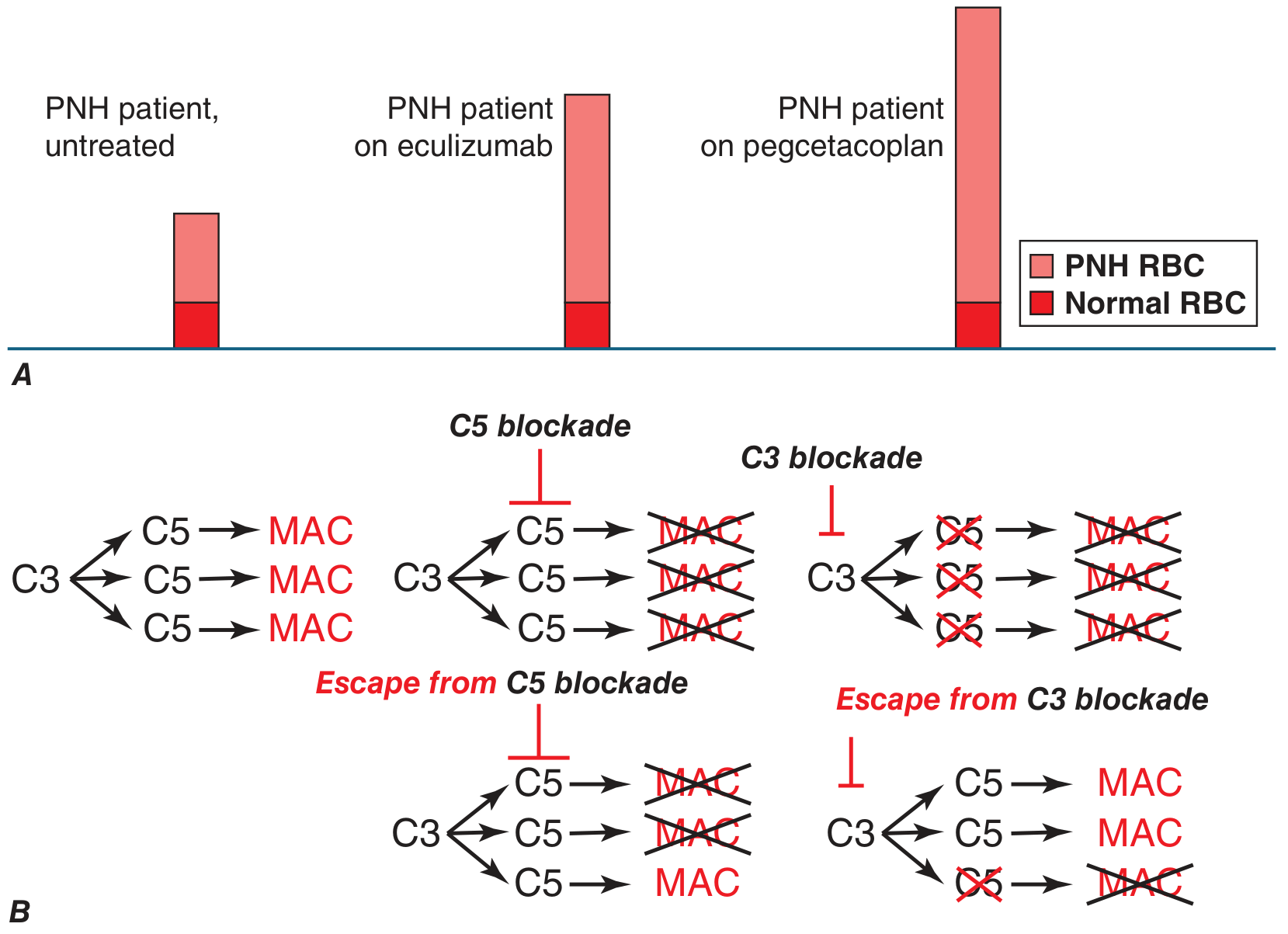
Current complement inhibitors:
| Drug | Target | Notes |
|---|---|---|
| Eculizumab | C5 | First approved; IV every 2 weeks; abolishes intravascular hemolysis; reduces thrombosis risk |
| Ravulizumab | C5 | Long-acting; IV every 8 weeks |
| Crovalimab | C5 | Subcutaneous; self-administered; 2026 systematic review confirms efficacy |
| Pegcetacoplan | C3 | Proximal inhibitor; prevents both intravascular AND extravascular hemolysis; corrects anemia more completely |
| Iptacopan | Factor B (alternative pathway) | Oral; targets upstream |
Key limitation of C5 inhibitors: PNH red cells, now protected from MAC lysis, still accumulate C3b fragments on their surface → become opsonized → iatrogenic extravascular hemolysis (Coombs test becomes positive). Pegcetacoplan, acting at C3, prevents this.
Breakthrough hemolysis risk: Incomplete C5 blockade → some MAC forms. With C3 blockade, if incomplete, the downstream enzymatic cascade amplifies - potentially more severe breakthrough. Clinicians must maintain adequate drug levels.
Meningococcal Vaccination - Mandatory
All patients on complement inhibitors must be vaccinated against Neisseria meningitidis (and should receive prophylactic antibiotics) because blockade of the terminal complement pathway removes a key defense against encapsulated organisms, especially meningococcus.
Allogeneic BMT (Stem Cell Transplant)
The only curative option. Reserved for:
- Severe aplastic anemia with PNH
- Refractory disease
- Significant cytopenias
- Carries nonnegligible procedure-related risks
Supportive Care
- Iron supplementation (for hemosiderinuria-related deficiency)
- Folate supplementation
- Anticoagulation for thrombosis (not clearly effective prophylactically in untreated patients; complement inhibitors reduce thrombosis risk more effectively)
- Transfusions when needed (washed RBCs preferred to avoid additional complement triggers)
- Corticosteroids may reduce some hemolytic episodes
Key Associations
- Aplastic anemia - bidirectional; PNH may evolve from AA; AA may follow PNH; autoimmune basis explains the link
- MDS - small subclinical PNH clones in 15-20% of MDS
- Budd-Chiari syndrome - think PNH in any young patient with hepatic vein thrombosis
- Iron deficiency with hemolytic markers - unusual combination that should raise PNH suspicion
Summary Mnemonics
PNH = PIGA mutation → No GPI → No CD55/CD59 → MAC attacks RBCs
Classic triad: Hemolysis + Thrombosis + Cytopenias
Remember: Coombs negative (unlike autoimmune hemolytic anemia), hemosiderinuria always present, nocturnal hemoglobinuria in minority only.
Sources:
- Robbins & Kumar Basic Pathology, p. 605-606
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 606
- Goldman-Cecil Medicine, Ch. 146
- Harrison's Principles of Internal Medicine 22E (2025), p. 848-849
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 693, 798
Recent literature: A 2026 systematic review and meta-analysis (PMID 41299829) confirmed the efficacy and safety of crovalimab (subcutaneous C5 inhibitor) in PNH, supporting its use as an alternative to IV anti-C5 therapy. A 2025 systematic review (PMID 40244439) examined PNH in pregnancy, where eculizumab is considered safe and is the treatment of choice given the high thrombotic risk.
This is a shared conversation. Sign in to Orris to start your own chat.