Pathogenesis of uremia and uremic coma. Pathogenic therapy of uremic
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
uremic encephalopathy pathogenesis treatment
Reading File
Loading Image
I now have all the material needed to compose a thorough, well-structured answer. Here is the comprehensive response:
Pathogenesis of Uremia, Uremic Coma, and Pathogenic Therapy
1. What Is Uremia?
Uremia (from Latin urina = urine, haemia = blood) is the clinical and biochemical syndrome that results from the accumulation of nitrogenous waste products and other solutes that are normally cleared by the kidneys. It occurs in both acute kidney injury (AKI) and end-stage kidney disease (ESKD). The term encompasses far more than just elevated BUN and creatinine - it reflects a multi-organ toxic state driven by retained solutes, hormonal dysregulation, and metabolic derangement.
2. Pathogenesis of Uremia
2.1 Uremic Toxins
The European Uremic Toxin (EUTox) database lists over 100 retained solutes in uremia. They are classified by size and protein binding:
| Category | Examples | Key Effects |
|---|---|---|
| Small water-soluble (<500 Da) | Urea, creatinine, guanidines, uric acid | Neurological, cardiovascular |
| Middle molecules (500-60,000 Da) | Beta-2-microglobulin, parathyroid hormone (PTH), cytokines | Neuropathy, amyloidosis |
| Protein-bound solutes | Indoxyl sulfate, p-cresyl sulfate | Endothelial damage, CKD progression |
Importantly, urea itself is NOT the primary toxin - urea infusions do not reproduce uremic symptoms, and hemodialysis reverses the syndrome even when urea is maintained in the dialysate. The level of BUN and creatinine correlates only loosely with symptom severity. - Plum and Posner's Diagnosis and Treatment of Stupor and Coma, p. 444
2.2 Guanidino Compounds - Central Neurotoxins
Guanidino compounds (guanidinosuccinic acid, methylguanidine, homoarginine, creatinine) are elevated 100-fold in uremic brain tissue and CSF. Their mechanism of neurotoxicity:
- They antagonize GABA-A receptors (reducing inhibitory tone)
- They act as agonists on NMDA glutamate receptors (enhancing excitatory tone)
- They induce seizures via calcium channel modulation
- Levels of guanidinouscinic acid and methylguanidine are increased 100-fold in uremic CSF
This imbalance of excitatory vs. inhibitory neurotransmission is central to uremic encephalopathy. - Comprehensive Clinical Nephrology, 7th Ed., p. 1197; Brenner & Rector's The Kidney, p. 2551
2.3 Blood-Brain Barrier Disruption and Brain Edema
- Kidney injury activates inflammatory cytokines that cross or alter the blood-brain barrier (BBB)
- Uremic solute retention triggers vascular permeability in the brain
- Diffusion-weighted MRI findings confirm brain edema in uremic encephalopathy
- Studies show increased brain inflammation in uremia (distinct from hepatic encephalopathy, where the mechanism is primarily ammonia-driven)
2.4 Calcium and Secondary Hyperparathyroidism
- Brain calcium is elevated in uremia
- PTH has direct CNS effects - it alters neuronal calcium transporters
- High PTH raises alkaline phosphatase in the brain, which promotes binding of tau protein to muscarinic receptors in the hippocampus, causing neuronal calcium overload and cell death
- In human studies, both cognitive function and EEG normalize after parathyroidectomy - Comprehensive Clinical Nephrology, 7th Ed.; Brenner & Rector's The Kidney
2.5 Neurotransmitter and Monoamine Dysregulation
- Depletion of norepinephrine and suppression of central dopamine - linked to impaired motor activity
- Elevated serotonin (from high tryptophan entry across BBB) - contributes to anorexia
- Elevated NPY, reduced neuropeptide signalling disrupt appetite regulation
- Elevated 3-hydroxykinurenine (neurotoxic tryptophan metabolite) accumulates in striatum and medulla
2.6 Oxidative Stress
- Upregulation of NADPH oxidase and downregulation of superoxide dismutase in the uremic brain
- Results in nitration of brain proteins and oxidative myelin damage
- Dialysis itself generates additional oxidative stress through blood-membrane interaction
2.7 Asymmetric Dimethylarginine (ADMA)
- ADMA is elevated in CKD
- Inhibits endothelial nitric oxide synthase (eNOS), impairing cerebrovascular regulation
- Correlates with cerebrovascular complications in uremia
2.8 Gut Microbiome-Derived Toxins
- Indoxyl sulfate (from tryptophan via gut bacteria) impairs BBB integrity
- Metabolites of phenylalanine, benzoate, and glutamate metabolism from gut flora are linked with cognitive impairment in dialysis patients
2.9 Drug Accumulation
- Meperidine (pethidine): normeperidine accumulates, causes seizures - contraindicated in CKD
- Morphine: active metabolites (morphine-6-glucuronide) accumulate
- Cimetidine and acyclovir metabolites increase due to inhibition of organic anion transporter 3 (OAT3)
- Cephalosporins can cause delirium, asterixis, myoclonus, and non-convulsive status epilepticus in uremic patients
2.10 Metabolic Derangements
Uremia is accompanied by:
- Metabolic acidosis with respiratory compensation (Kussmaul breathing)
- Hyperkalemia
- Hyperphosphatemia and hypocalcemia
- Hyponatremia (from water retention)
- Anemia (decreased EPO production)
Note: Systemic acidosis does NOT cause the CNS symptoms of uremia directly - the BBB buffers central pH, and correction of blood pH does not improve cerebral symptoms.
3. Pathogenesis of Uremic Coma (Uremic Encephalopathy)
Uremic encephalopathy (UE) is a metabolic encephalopathy representing the most severe manifestation of the uremic syndrome on the nervous system.
3.1 Spectrum of Severity
Early Encephalopathy:
- Mood swings, emotional lability
- Fatigue, drowsiness, restlessness
- Reduced attention span
- Fine action tremor
Intermediate:
- Memory and cognitive deficits
- Reversal of sleep-wake cycle
- Asterixis (intermittent loss of antigravity muscle tone - "negative myoclonus")
- Multifocal myoclonus
- Slurred speech, dysarthria
Late/Severe Encephalopathy (Uremic Coma):
- Delirium - florid with hallucinations and agitation (acute onset) OR quiet apathy (chronic onset)
- Psychosis
- Hemiparesis (shifting from side to side - a hallmark)
- Tetany (especially with alkalinisation)
- Generalized convulsions and non-convulsive status epilepticus
- Stupor and coma
"The characteristic combination of dulled consciousness, hyperpnea, and motor hyperactivity should immediately give high suspicion to the diagnosis." - Plum & Posner, p. 444
3.2 Neurophysiology of Uremic Coma
At the cellular level:
- Decreased cerebral metabolic rate and oxygen consumption
- Reduced glycolysis and energy utilization (though cerebral high-energy phosphates are maintained)
- Decreased sodium/potassium flux and depressed Na-K-ATPase activity
- These are effects, not primary causes - the initiating insult is toxic
EEG findings: Generalized slowing (theta and delta waves), correlating with degree of azotemia. Non-specific but helpful.
Brain morphology: No consistent abnormality on neuropathology. Uremia uncomplicated by hypertension does NOT cause cerebral edema. MRI may show cortical and subcortical T2 hyperintensities, particularly in occipital lobes and basal ganglia (reversible with dialysis).
3.3 Diagnostic Points
- BUN commonly >200 mg/dL in severe UE, but correlation is poor (some patients have severe delirium at BUN 48 mg/dL; others are symptom-free at BUN >200)
- CSF: modest pleocytosis (<25 cells/μL), elevated protein (<100 mg/dL), elevated pressure (>160 mmH₂O) - aseptic meningitis pattern
- Pupillary and oculomotor functions are not disturbed (unlike structural coma)
- Differential: hypertensive encephalopathy, dialysis disequilibrium, water intoxication, drug toxicity (cephalosporins, opioids), metabolic acidosis from other causes
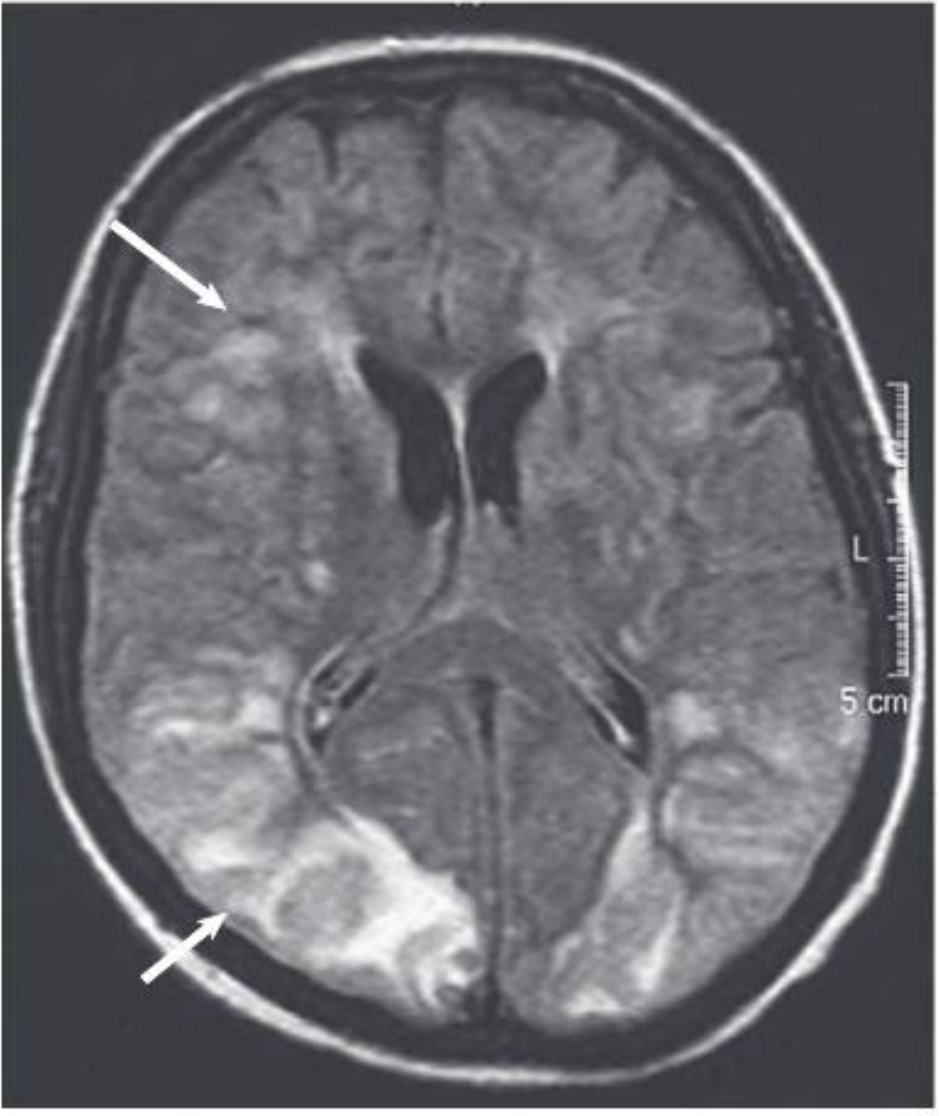
MRI (T2-FLAIR) from a 40-year-old with uremic encephalopathy: extensive hyperintense lesions in cortical/subcortical occipital areas and focal frontal white matter (arrows). These changes reversed within 2 weeks of initiating dialysis. - Comprehensive Clinical Nephrology, 7th Ed., Fig. 89.1
4. Pathogenic Therapy of Uremia
"Pathogenic therapy" means treatment directed at the underlying mechanism of disease, not just symptomatic relief. The primary pathogenic approach in uremia is to remove the retained toxic solutes.
4.1 Kidney Replacement Therapy (KRT) - Primary Pathogenic Treatment
This is the cornerstone - it removes water-soluble uremic toxins, restores fluid and electrolyte balance, and corrects acidosis.
Indications for initiating KRT:
- Uremic encephalopathy or coma
- Uremic pericarditis
- Anorexia/nausea/vomiting not attributable to other causes
- Refractory hyperkalemia
- Refractory volume overload
- Malnutrition from uremic anorexia
"Prompt treatment of uremic encephalopathy with initiation or intensification of renal replacement therapy is indicated. Resolution of symptoms typically occurs within days." - Brenner & Rector's The Kidney
Modalities:
| Modality | Notes |
|---|---|
| Intermittent hemodialysis (IHD) | Most common; efficient small solute removal |
| Peritoneal dialysis (PD) | Better for hemodynamically unstable; can be used in ICU |
| Continuous veno-venous hemofiltration (CVVHF/CVVHD) | Preferred in unstable patients, AKI in ICU; avoids rapid osmolar shifts |
| Kidney transplantation | Only complete cure - restores endocrine functions dialysis cannot; encephalopathy resolves within days post-transplant |
Note on delayed recovery: Neurological recovery does not always immediately follow dialysis. Patients may remain comatose for days after biochemical normalization. This does NOT indicate permanent brain damage - full neurological recovery follows in most cases.
4.2 Prevention and Treatment of Dialysis Disequilibrium Syndrome
A complication of the pathogenic treatment itself: rapid removal of urea creates an osmotic gradient where the brain (with its idiogenic osmoles) becomes hyperosmolar relative to blood, drawing in water and causing cerebral edema.
Prevention:
- Slower, lower-efficiency initial dialysis sessions
- Addition of osmotically active solutes (mannitol, urea, glycerol, sodium) to dialysate to maintain blood osmolarity
- Increasing dialysate sodium concentration
- Earlier initiation of dialysis before severe azotemia develops
4.3 Correction of Secondary Hyperparathyroidism
Directed at the PTH-mediated calcium accumulation in the brain:
- Phosphate binders (calcium carbonate, sevelamer, lanthanum)
- Vitamin D analogues (calcitriol, paricalcitol) to suppress PTH
- Calcimimetics (cinacalcet) to suppress PTH secretion
- Parathyroidectomy in refractory cases - documented improvement in cognitive function and EEG normalization
4.4 Correction of Anemia (Erythropoiesis-Stimulating Agents)
Anemia contributes to CNS dysfunction through cerebral hypoxia:
- Recombinant erythropoietin (rHuEPO) therapy in dialysis patients is associated with improved cognitive function and decreased EEG slowing
- Caution: too rapid correction of anemia can precipitate seizures
4.5 Dietary Protein Restriction
- Reduces generation of nitrogenous waste products (urea, guanidines)
- Delays onset of uremic symptoms
- Typical: 0.6-0.8 g/kg/day protein (with essential amino acid supplementation if very restrictive)
- Risk of malnutrition limits utility as a long-term sole strategy
- Should be a bridge to KRT, not a substitute
4.6 Correction of Metabolic Derangements
| Derangement | Approach |
|---|---|
| Metabolic acidosis | Oral sodium bicarbonate supplementation (aim serum bicarbonate >22 mmol/L) |
| Hyperkalemia | Dietary restriction, potassium binders (patiromer, sodium zirconium cyclosilicate), dialysis |
| Hyperphosphatemia | Dietary restriction, phosphate binders |
| Hyponatremia | Fluid restriction; dialysate sodium adjustment |
| Hypertension | ACE inhibitors/ARBs (also nephroprotective), amlodipine |
4.7 Gut Microbiome-Targeted Therapy (Emerging)
Since gut-derived uremic toxins (indoxyl sulfate, p-cresyl sulfate) are protein-bound and poorly dialyzable:
- Prebiotics and probiotics to alter gut flora composition
- Dietary interventions to reduce fermentation of aromatic amino acids
- Activated charcoal (AST-120) to adsorb intestinal precursors - studied in CKD progression [reviewed in Andrews et al., JASN 2025, PMID 40009460]
4.8 Treatment of Uremic Coma - Acute Management
- Establish/intensify dialysis immediately - IV access, urgent hemodialysis if AKI; initiate KRT if not already started
- Protect airway - intubation if GCS ≤8 or if rapidly deteriorating
- Correct electrolytes - treat hyperkalemia emergently (calcium gluconate, insulin-dextrose, sodium bicarbonate)
- Treat seizures - with agents that do not accumulate in renal failure; avoid meperidine; levetiracetam dose-adjusted for GFR; benzodiazepines for acute seizure control
- Correct acidosis - IV sodium bicarbonate if pH <7.1 or bicarbonate <15
- Treat hypertensive encephalopathy if co-existing - IV labetalol, nicardipine
- Thiamine supplementation - patients on chronic dialysis are at risk for Wernicke's encephalopathy; IV thiamine 100 mg before any glucose
- Review all medications - remove renally-cleared neurotoxins (cephalosporins, meperidine, morphine accumulation)
Summary Table
| Mechanism | Key Pathogen | Pathogenic Therapy |
|---|---|---|
| Uremic solute retention | Guanidines, indoxyl sulfate | Dialysis, dietary protein restriction |
| Secondary hyperparathyroidism | PTH-mediated brain Ca²⁺ accumulation | Vitamin D, calcimimetics, parathyroidectomy |
| Neurotransmitter imbalance | GABA↓, NMDA hyperactivation | Dialysis (removes guanidines) |
| Anemia | Cerebral hypoxia | Erythropoietin |
| Gut toxins | Protein-bound solutes | Gut microbiome modification, AST-120 |
| Metabolic acidosis | CNS acid load | Bicarbonate, dialysis |
| Oxidative stress | NADPH oxidase activation | Dialysis, antioxidants (investigational) |
Sources:
- Comprehensive Clinical Nephrology, 7th Edition, Chapter 89: Uremic Encephalopathy
- Plum and Posner's Diagnosis and Treatment of Stupor and Coma, pp. 444-446
- Brenner and Rector's The Kidney, pp. 2550-2551, 4618-4641
- Harrison's Principles of Internal Medicine 22E (2025), Chapter on CKD/RRT
- Andrews et al., "Uremic Toxins, CKD, and Cognitive Dysfunction," JASN 2025 (PMID 40009460)
This is a shared conversation. Sign in to Orris to start your own chat.