What are dna oncogenic virus in pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from Robbins Pathology. Let me compile the full answer.
DNA Oncogenic Viruses in Pathology
(Based primarily on Robbins, Cotran & Kumar - Pathologic Basis of Disease and Schwartz's Principles of Surgery)
Overview
DNA oncogenic viruses differ fundamentally from RNA oncogenic viruses (retroviruses). Their oncogenes are viral in origin (not derived from cellular proto-oncogenes). In permissive (productive) hosts, infection causes a lytic cycle ending in cell death and viral release. In nonpermissive cells, viral DNA integrates into the host chromosome; early viral genes are persistently expressed, transforming cells into a neoplastic state. The key shared mechanism is binding of viral oncoproteins to tumor-suppressor proteins p53 and RB.
The five major DNA oncogenic viruses implicated in human cancer are:
- Human Papillomavirus (HPV)
- Epstein-Barr Virus (EBV)
- Hepatitis B Virus (HBV)
- Merkel Cell Polyomavirus (MCPyV)
- Human Herpesvirus 8 (HHV-8 / Kaposi Sarcoma Herpesvirus)
1. Human Papillomavirus (HPV)
Type: DNA virus (double-stranded circular DNA)
High-risk types: 16, 18, 31 (carcinogenic) | Low-risk types: 6, 11 (benign warts)
Associated cancers:
- Cervical carcinoma (most important)
- Anal, vulvar, vaginal, penile carcinomas
- Oropharyngeal carcinoma (tonsil, base of tongue)
Mechanism of Transformation
In benign warts, HPV DNA is maintained as a non-integrated episome. In cancers, the HPV genome integrates into the host genome, interrupting the E1/E2 open-reading frame - this destroys the E2 repressor, leading to markedly increased expression of E6 and E7 oncoproteins.
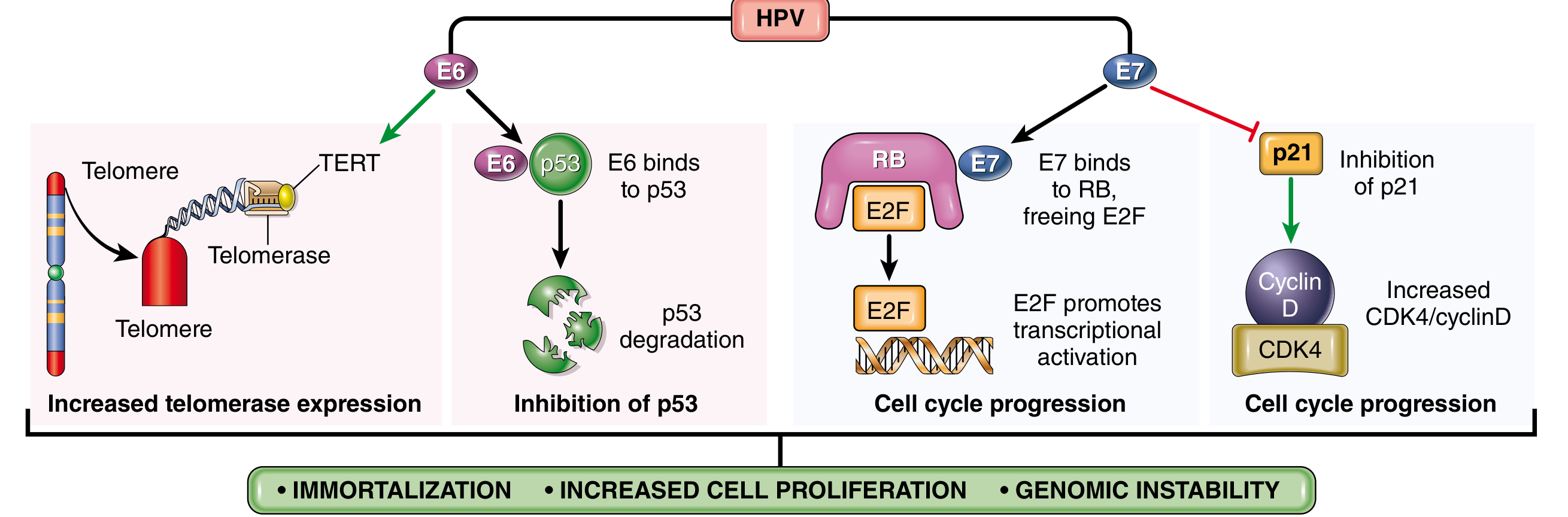
Fig. 7.44 from Robbins Pathology - Transforming effects of HPV E6 and E7 proteins
E6 oncoprotein actions:
- Binds to and mediates degradation of p53 (prevents DNA repair and apoptosis)
- Stimulates expression of TERT (telomerase reverse transcriptase) - promotes immortalization
- High-risk HPV E6 has much higher affinity for p53 than low-risk E6
E7 oncoprotein actions:
- Binds RB protein and displaces E2F transcription factors, pushing cells through the G1/S checkpoint
- Inactivates CDK inhibitors p21 and p27
- Binds and activates cyclins A and E (in high-risk types 16, 18, 31)
- High-risk E7 has higher affinity for RB than low-risk E7
Net result: Immortalization + increased cell proliferation + genomic instability
HPV infection alone is not sufficient for cancer - other genetic hits (e.g., mutant RAS), smoking, HIV co-infection, and immune failure are needed, typically over several decades.
2. Epstein-Barr Virus (EBV)
Type: Herpesvirus family (dsDNA)
Associated cancers:
- Burkitt lymphoma (first human tumor linked to EBV)
- Hodgkin lymphoma
- Nasopharyngeal carcinoma (100% EBV-associated worldwide)
- Immunosuppression-related B-cell lymphomas
- EBV-positive gastric carcinoma, thymic carcinoma
Mechanism
EBV uses its surface glycoproteins to bind CD21 (complement receptor) on B cells, establishing latent infection - no lytic replication, cells are not killed. Instead, EBV genes immortalize B cells by hijacking normal signaling pathways:
- LMP1 (Latent Membrane Protein-1): Acts as a constitutively active CD40 receptor. Activates NF-κB and JAK/STAT signaling, promotes B-cell survival and proliferation, and prevents apoptosis via BCL2 activation. In nasopharyngeal carcinoma, LMP1 also upregulates VEGF, FGF2, MMP9, and COX2.
- EBNA2: Encodes a nuclear protein that activates expression of several cellular and viral genes, including the proto-oncogene MYC.
Burkitt Lymphoma
EBV acts as a polyclonal B-cell mitogen, setting the stage for the acquisition of the t(8;14) translocation (MYC dysregulation) and other mutations. EBV is not directly oncogenic here - it expands the pool of proliferating B cells, increasing the probability of secondary mutations. Chronic malaria co-infection (in endemic areas) favors persistent EBV infection, contributing to the geographic pattern of endemic Burkitt lymphoma.
Nasopharyngeal Carcinoma
All nasopharyngeal carcinomas worldwide contain EBV (clonal viral genome, indicating infection preceded tumor). LMP1 is expressed and tumor cells often express PD-L1, enabling immune evasion.
3. Hepatitis B Virus (HBV)
Type: Partially double-stranded DNA virus (Hepadnavirus)
Associated cancer: Hepatocellular carcinoma (HCC) - HBV/HCV together account for 70-85% of HCCs worldwide; highest incidence in Far East and Africa.
Mechanism
Unlike HPV and EBV, no viral oncogene has been identified in HBV. The proposed mechanisms include:
- Chronic liver injury + regenerative hyperplasia - repeated cycles of hepatocyte death and regeneration from a persistent inflammatory response increase the probability of accumulating mutations
- HBV X protein (HBx): Transactivates various growth-regulatory genes and may impair p53 function
- Viral DNA integrates randomly into the host genome, potentially causing insertional mutagenesis (though this is random and not the primary mechanism)
- Cirrhosis acts as a permissive background for neoplastic transformation
HBV vaccination in childhood has already reduced liver cancer incidence in East Asia.
4. Merkel Cell Polyomavirus (MCPyV)
Type: Small dsDNA virus (Polyomaviridae)
Associated cancer: Merkel cell carcinoma - a rare but aggressive neuroendocrine skin cancer.
Mechanism
- Viral DNA integrates into the host genome in a clonal fashion (before tumor development)
- Expresses T antigens (large T antigen) that bind and inactivate RB and p53, similar to other polyomaviruses (e.g., SV40)
- Truncating mutations in the large T antigen prevent viral replication but preserve transforming capacity
5. Human Herpesvirus 8 (HHV-8 / KSHV)
Type: Gamma-herpesvirus (dsDNA)
Associated cancers:
- Kaposi sarcoma (classic, endemic, AIDS-related, and transplant-related forms)
- Primary effusion lymphoma
- Multicentric Castleman disease
Mechanism
- HHV-8 encodes viral homologs of cellular genes, including v-cyclin D, v-FLIP (inhibits apoptosis), and v-IL-6
- v-cyclin D binds CDK6 and drives cells through the G1/S checkpoint
- v-FLIP activates NF-κB, promoting cell survival
- Viral GPCR (vGPCR) activates VEGF and angiogenic pathways, contributing to the vascular nature of Kaposi sarcoma
Summary Table
| Virus | Type | Key Oncogenic Mechanism | Associated Cancer(s) |
|---|---|---|---|
| HPV 16/18 | dsDNA | E6 degrades p53; E7 inactivates RB; TERT activation | Cervical, oropharyngeal, anogenital carcinomas |
| EBV | dsDNA (Herpes) | LMP1 mimics CD40 (NF-κB, JAK/STAT); EBNA2 activates MYC | Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal carcinoma |
| HBV | Partial dsDNA | Chronic inflammation + regeneration; HBx protein | Hepatocellular carcinoma |
| MCPyV | dsDNA | T antigen inactivates RB and p53 | Merkel cell carcinoma |
| HHV-8 | dsDNA (Herpes) | v-cyclin D, v-FLIP (NF-κB), vGPCR/VEGF | Kaposi sarcoma, primary effusion lymphoma |
Key Shared Concept
"The binding of viral oncoproteins to cellular tumor-suppressor proteins p53 and RB is fundamental to the carcinogenesis induced by most DNA viruses."
- Robbins, Cotran & Kumar - Pathologic Basis of Disease
Viral carcinogenesis is always a multistep process - the virus creates a permissive environment and early genetic changes, but additional hits (genetic mutations, immune suppression, environmental co-factors) are required for full malignant transformation. This explains why most infected individuals never develop cancer, and why cancers typically arise decades after initial infection.
This is a shared conversation. Sign in to Orris to start your own chat.