Combine micro patho and pharma of tuberculosis
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the integrated response.
Tuberculosis: Microbiology, Pathology & Pharmacology — Integrated
PART 1: MICROBIOLOGY
The Organism — Mycobacterium tuberculosis
M. tuberculosis (MTB) is a non-motile, non-spore-forming, slightly curved, obligate aerobe. It is weakly Gram-positive but the defining characteristic is acid-fastness:
- The cell wall contains mycolic acids (long-chain fatty acids, C60-C90) and lipoarabinomannan (LAM), a lipid-polysaccharide complex extending from the plasma membrane to the surface
- Lipids make up >60% of total cell wall mass — creating a "waxy coat" that is impenetrable and hydrophobic
- Once stained (Ziehl-Neelsen: carbol-fuchsin + heat), the stain is fast even after decolorization with acid-alcohol → retained red color against a blue background
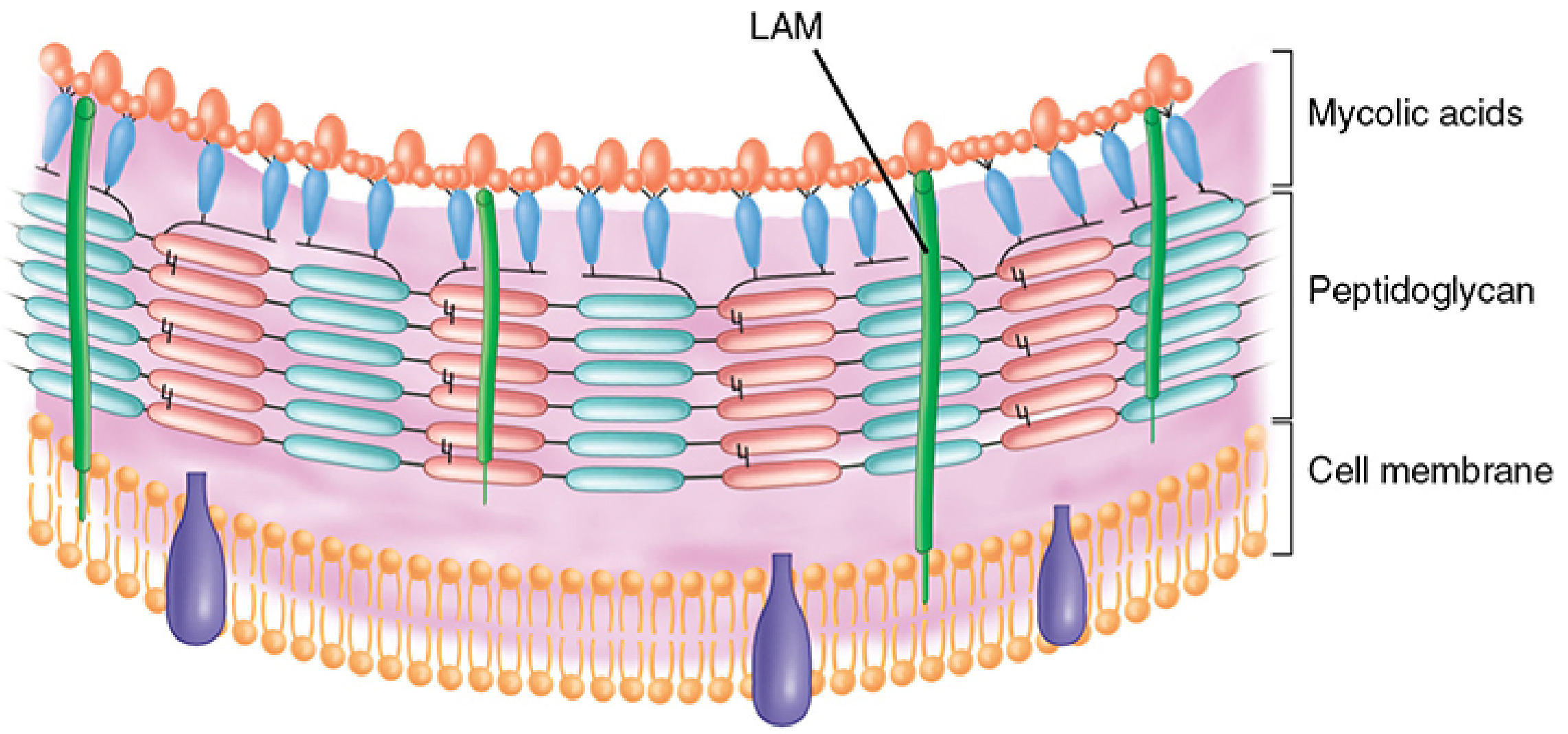
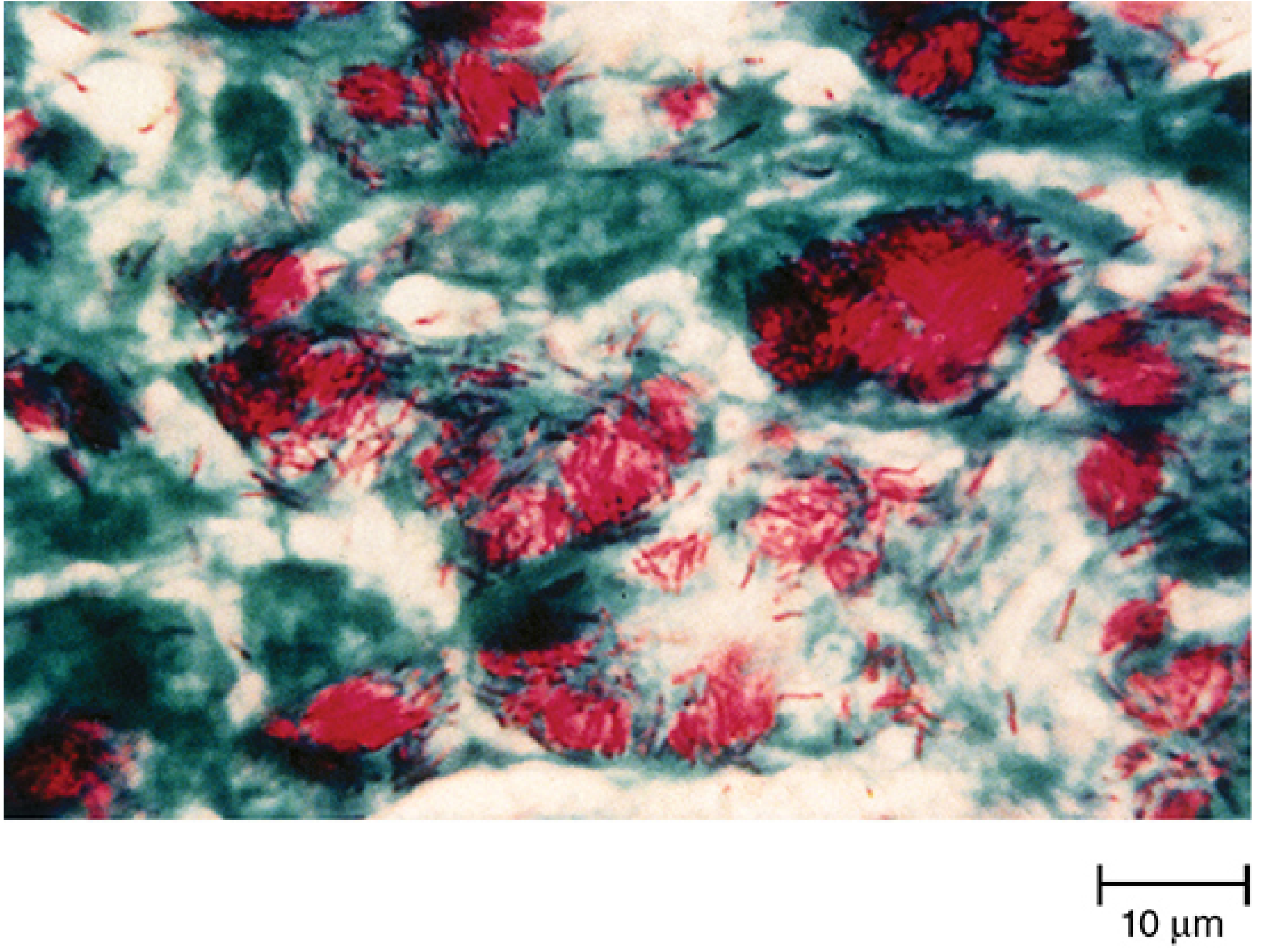
Growth characteristics: Strict aerobe, grows in 5-10% CO2, optimal pH 6.5-6.8, extremely slow doubling time (15-20 hours). Does NOT grow on standard bacteriologic media; requires enriched media (Lowenstein-Jensen, Middlebrook 7H10).
PART 2: PATHOGENESIS & PATHOLOGY
Transmission
MTB is transmitted via aerosolized droplet nuclei (1-5 µm), small enough to reach the alveoli. Coughing is promoted by sulfolipid in the mycobacterial cell envelope. Cavitary lesions (containing 10⁷-10⁹ bacilli) are the most infectious source. One infectious individual infects 3-10 people per year; in household settings, 25-50% of contacts become infected.
Stage 1 — Initial Infection and Macrophage Evasion
When MTB bacilli reach the distal alveoli, they are phagocytosed by alveolar macrophages. Normally macrophages kill pathogens, but MTB has evolved multiple mechanisms to subvert this:
| Mechanism | Effect |
|---|---|
| Inhibits phagosome-lysosome fusion | Survives inside the phagosome |
| Scavenges reactive oxygen species | Evades oxidative killing |
| Secretes virulence factors (sulfolipids, cord factor = trehalose dimycolate) | Inhibits macrophage activation |
| Modulates host signaling pathways | Blocks apoptosis of infected macrophage |
Monocyte-derived macrophages, interstitial macrophages, and neutrophils are recruited but also fail to control MTB replication. Infected dendritic cells travel to draining lymph nodes to prime T cells.
Stage 2 — Adaptive Immunity and Granuloma Formation
The interval from initial infection to a positive tuberculin skin test (TST) / IGRA is 2-8 weeks. During this window, MTB grows relatively unhindered.
Once the adaptive response mounts, T cells, B cells, and activated macrophages form the granuloma - the histopathologic hallmark of MTB infection:
Granuloma structure:
- Central zone: caseous necrosis (cheese-like) - caused by delayed-type hypersensitivity (DTH) response
- Middle zone: epithelioid macrophages (activated macrophages with abundant cytoplasm) and Langhans-type multinucleated giant cells (macrophage fusion)
- Peripheral zone: CD4+ and CD8+ T lymphocytes + fibrous collar
Key cytokines:
- IFN-γ (from CD4 T cells) → activates macrophages for mycobactericidal activity
- TNF-α → critical for granuloma formation and maintenance
- IL-12 → drives Th1 differentiation
Patients with CD4 T-cell dysfunction (HIV) or TNF-α inhibitor therapy fail to form well-organized granulomas → high risk for active TB and disseminated infection.
Stage 3 — Primary TB (Ghon Complex)
In most immunocompetent individuals, primary infection is asymptomatic and self-limited:
- Subpleural focus of infection in the lower lobe or mid-zone → Ghon focus
- Ghon focus + ipsilateral hilar lymphadenopathy = Ghon complex
- Over time, granulomas calcify (Ranke complex on CXR)
- MTB enters a latent state (dormant in macrophages) — risk of reactivation persists for life
~5-10% of immunocompetent infected individuals will develop active TB at some point. HIV co-infection increases this to ~10% per year.
Stage 4 — Reactivation (Post-Primary / Secondary TB)
Occurs when latent MTB reactivates, typically in the apical and posterior segments of the upper lobes (high O₂ tension). Features include:
- Cavitation — necrosis with liquefaction, cavity walls show MTB proliferating up to 10⁹ bacilli/mL
- Bronchial spread — coughing seeds other lung segments
- Fibrosis → destroyed lung parenchyma replaced by fibrous tissue
Progressive pathological sequence: Exudative lesion → caseation → liquefaction → cavity formation → fibrosis/calcification
Extrapulmonary TB
MTB disseminates via lymphatics and bloodstream from primary focus:
| Site | Features |
|---|---|
| Lymph nodes | Most common extrapulmonary site; "scrofula" (cervical nodes) |
| Pleura | Pleural effusion (exudative, lymphocyte-predominant) |
| CNS | Meningitis - basilar involvement; tuberculomas |
| Spine (Pott's disease) | Lower thoracic/upper lumbar vertebrae; paravertebral abscess; gibbus deformity |
| Genitourinary | Sterile pyuria; "putty kidney" calcification |
| Miliary TB | Hematogenous dissemination → millet-seed lesions in all organs |
| Pericardium | Constrictive pericarditis over time |
| GI tract | Terminal ileum most common; mimics Crohn's disease |
PART 3: PHARMACOLOGY
The goal of anti-TB treatment is to kill three populations of MTB simultaneously:
- Rapidly dividing extracellular bacilli (in cavities) → isoniazid, rifampin
- Slowly dividing intracellular bacilli (in macrophages) → pyrazinamide
- Dormant/persister bacilli → rifampin (sterilizing activity)
First-Line Drugs (RIPE / HRZE)
1. Isoniazid (INH / H)
| Property | Detail |
|---|---|
| Mechanism | Prodrug activated by KatG (mycobacterial catalase-peroxidase) → isonicotinoyl radical → inhibits InhA (enoyl-ACP reductase) and KasA → blocks mycolic acid synthesis → cell wall disruption |
| Additional effects | KatG also generates superoxide, H₂O₂, and NO radicals → mycobactericidal via oxidative damage |
| Activity | Bactericidal against actively dividing MTB; bacteriostatic against slow-growers |
| Dose | 300 mg/day (adults) |
| Resistance | Mutations in katG (most common - loss of activation) or inhA promoter (overexpression of target) |
| Key adverse effects | Hepatotoxicity (most serious - idiosyncratic, dose-related in high doses); peripheral neuropathy (due to pyridoxine/B6 depletion - give B6 prophylactically); drug-induced lupus; CNS effects |
| Special | The single most important TB drug; used alone for LTBI treatment (6-9 months) |
2. Rifampin (Rifampicin / R)
| Property | Detail |
|---|---|
| Mechanism | Inhibits bacterial DNA-dependent RNA polymerase (β-subunit, encoded by rpoB) → blocks mRNA synthesis → bactericidal |
| Activity | Bactericidal; also active against semi-dormant bacteria ("sterilizing" activity) |
| Dose | 600 mg/day |
| Resistance | Mutations in rpoB gene (>95% of rifampin-resistant strains) |
| Key adverse effects | Hepatotoxicity; orange-red discoloration of body fluids (urine, tears, sweat); flu-like syndrome (with intermittent use); thrombocytopenia |
| Drug interactions | Potent inducer of CYP450 (3A4) → reduces levels of oral contraceptives, warfarin, antiretrovirals, methadone, and many other drugs |
| Special | Never use as monotherapy (rapid resistance development); part of the "sterilizing" pair with isoniazid |
3. Pyrazinamide (PZA / Z)
| Property | Detail |
|---|---|
| Mechanism | Prodrug converted by mycobacterial pyrazinamidase/nicotinamidase (PncA) to pyrazinoic acid (POA) → disrupts mycobacterial membrane potential and energy metabolism; inhibits fatty acid synthase I (FASI) → blocks mycolic acid synthesis; only active at acidic pH (pH 5.5) |
| Why it matters | Selectively kills slowly dividing intracellular bacilli within the acidic phagolysosomal environment → responsible for shortening therapy from 9 to 6 months |
| Dose | 25 mg/kg/day |
| Resistance | Mutations in pncA gene |
| Key adverse effects | Hepatotoxicity (most serious ADR); hyperuricemia (inhibits tubular secretion of urate → gout); arthralgia; avoid in pregnancy (insufficient safety data in US) |
4. Ethambutol (EMB / E)
| Property | Detail |
|---|---|
| Mechanism | Inhibits arabinosyl transferases (encoded by embCAB) → blocks polymerization of arabinoglycan (essential cell wall component) |
| Activity | Bacteriostatic (not bactericidal) at standard doses |
| Dose | 15-25 mg/kg/day |
| Resistance | Mutations in embB or overexpression of emb gene products |
| Key adverse effects | Retrobulbar (optic) neuritis → decreased visual acuity, red-green color blindness (dose-dependent, usually reversible if stopped promptly; monthly visual acuity monitoring required); accumulates in renal failure |
| Role | Included as the 4th drug to protect against isoniazid/rifampin resistance until susceptibility is known; does NOT shorten therapy |
Treatment Regimens
Drug-Susceptible TB (Standard 6-Month Regimen)
| Phase | Duration | Drugs | Rationale |
|---|---|---|---|
| Intensive | 2 months | HRZE (Isoniazid + Rifampin + Pyrazinamide + Ethambutol) | Kill rapidly dividing + intracellular bacilli; reduce bacterial burden rapidly |
| Continuation | 4 months | HR (Isoniazid + Rifampin) | Eliminate remaining persisters; prevent relapse |
| Total | 6 months | - | PZA in intensive phase is what allows 6 vs. 9 months |
Newer 4-Month Regimen (Rifapentine-based): Rifapentine + Moxifloxacin + Isoniazid + Pyrazinamide (intensive 8 weeks) → noninferior to standard 6-month regimen in drug-susceptible pulmonary TB in adults ≥12 years, ≥40 kg.
Latent TB Infection (LTBI) Treatment
- 9H: Isoniazid daily × 9 months (standard)
- 6H: Isoniazid daily × 6 months (equivalent in most settings)
- 3HP: Isoniazid + Rifapentine weekly × 12 doses (3 months) - noninferior, higher completion rate
Drug-Resistant TB
| Type | Definition | Treatment |
|---|---|---|
| MDR-TB | Resistant to isoniazid + rifampin | BPaLM: Bedaquiline + Pretomanid + Linezolid + Moxifloxacin × 6 months (preferred) |
| XDR-TB | MDR + resistant to fluoroquinolones + second-line injectables | BPaL (Bedaquiline + Pretomanid + Linezolid) - FDA approved |
Second-Line Drugs (Brief Overview)
| Drug | Mechanism | Key ADR |
|---|---|---|
| Bedaquiline | Inhibits mycobacterial ATP synthase (Fo subunit) | QT prolongation |
| Linezolid | 23S rRNA inhibitor → protein synthesis | Myelosuppression, peripheral neuropathy |
| Fluoroquinolones (Moxifloxacin, Levofloxacin) | DNA gyrase + topoisomerase IV inhibition | QT prolongation |
| Cycloserine | Inhibits alanine racemase → blocks peptidoglycan synthesis | Seizures, psychosis |
| Clofazimine | Membrane destabilization + reactive oxygen species → kills "persister" MTB | Orange skin discoloration |
| Aminoglycosides (Amikacin, Streptomycin) | 30S ribosomal inhibition → protein synthesis | Nephrotoxicity, ototoxicity |
| Ethionamide | Prodrug → inhibits InhA (same target as INH) | GI intolerance, hepatotoxicity |
PART 4: THE PHARMACOLOGY-PATHOLOGY LINK
This table shows how each drug's mechanism directly addresses a specific pathological population of MTB:
| TB Population | Location | pH | Growth State | Killed by |
|---|---|---|---|---|
| Rapidly multiplying bacilli | Cavity walls, extracellular | Neutral | Fast | Isoniazid, Rifampin |
| Intracellular bacilli | Macrophage phagolysosome | Acidic (~5.5) | Slow | Pyrazinamide |
| Semi-dormant / persister bacilli | Old granulomas, caseous lesions | Neutral-slightly acidic | Intermittent bursts | Rifampin |
| Cell wall synthesis (all phases) | Cell wall | Any | Any | Isoniazid (mycolic acids), Ethambutol (arabinoglycan) |
The reason four drugs are needed (not one) is that a single drug would kill only one population, and the surviving populations would develop resistance. Using HRZE simultaneously collapses all viable niches simultaneously.
Sources:
- Goldman-Cecil Medicine, 26e - Ch. 299 (Tuberculosis: Pathobiology, Clinical Manifestations, Diagnosis, Treatment)
- Goodman & Gilman's Pharmacological Basis of Therapeutics, 14e - Ch. 65 (Antimycobacterial Agents)
- Katzung's Basic & Clinical Pharmacology, 16e - Ch. 47 (Drugs Used in Tuberculosis)
- Harrison's Principles of Internal Medicine, 22e - First-Line Antituberculosis Drugs
- Sherris & Ryan's Medical Microbiology, 8e - Ch. 27 (Mycobacteria)
- Medical Microbiology 9e - Treatment, Prevention, and Control
This is a shared conversation. Sign in to Orris to start your own chat.