Mi
It looks like your message got cut off! Could you finish your thought? What would you like help with?
Myocardial infarction
Asking for Preferences
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I have excellent material to compile a comprehensive answer. Here is the full response:
Myocardial Infarction (MI)
1. Definition & Epidemiology
A myocardial infarction is the death of cardiac muscle due to prolonged ischemia. In the United States alone, roughly 800,000 individuals experience an MI each year (nearly one MI every 40 seconds), causing almost 400,000 deaths annually. While MIs can occur at virtually any age, 10% occur in people younger than 40 years, and 45% in people younger than 65 years. Male sex increases relative risk through middle age; women gain relative protection during reproductive years from estrogen. After menopause, the risk in women rises sharply, making ischemic heart disease (IHD) the most common cause of death in older females.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
2. Pathogenesis
Coronary Arterial Occlusion (Typical - 90% of cases)
The sequence underlying most MIs follows these steps:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood.
- Platelets adhere, aggregate, and are activated, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Activation of coagulation by tissue factor adds to the growing thrombus.
- Within minutes, the thrombus completely occludes the coronary artery lumen.
When angiography is performed within 4 hours of MI onset, thrombotic occlusion is found in almost 90% of cases. Importantly, these thrombi are usually at a site that did not previously have a critical (>70%) fixed stenosis.
Atypical Causes (~10% of cases)
- Vasospasm (with or without atherosclerosis) - e.g., cocaine, ephedrine use
- Emboli from mural thrombus, infective endocarditis vegetations, or patent foramen ovale
- Vasculitis, sickle cell disease, amyloid deposition in vessel walls
- Robbins, Cotran & Kumar, p. 511
Myocardial Response to Ischemia
| Feature | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
The first biochemical consequence of ischemia is cessation of aerobic metabolism within seconds, leading to inadequate production of high-energy phosphates and accumulation of lactic acid. Contractility ceases within ~1 minute. Irreversible necrosis begins after 20-40 minutes of severe ischemia (flow ≤10% of normal), and becomes complete by 6-12 hours. Subendocardial muscle is especially susceptible because it is the last area to receive blood from epicardial vessels and faces high intramural pressures that impede inflow.
- Robbins, Cotran & Kumar, p. 512
- Guyton and Hall Textbook of Medical Physiology, p. 271
3. Patterns of Infarction
Two major patterns are recognized:
- Transmural infarction: Full-thickness necrosis of the ventricular wall; caused by complete occlusion of an epicardial vessel. Associated with STEMI on ECG.
- Subendocardial (non-transmural) infarction: Necrosis limited to the inner third of the ventricular wall; occurs with transient/partial obstruction or global hypotension. Associated with NSTEMI/UA on ECG.
Coronary Artery Territory
| Artery | Frequency | Area Infarcted |
|---|---|---|
| Left anterior descending (LAD) | 40-50% | Anterior LV wall, anterior 2/3 of interventricular septum, apex |
| Right coronary artery (RCA) | 30-40% | Inferior/posterior LV wall, posterior 1/3 of septum, posterior RV |
| Left circumflex (LCX) | 15-20% | Lateral wall of LV |
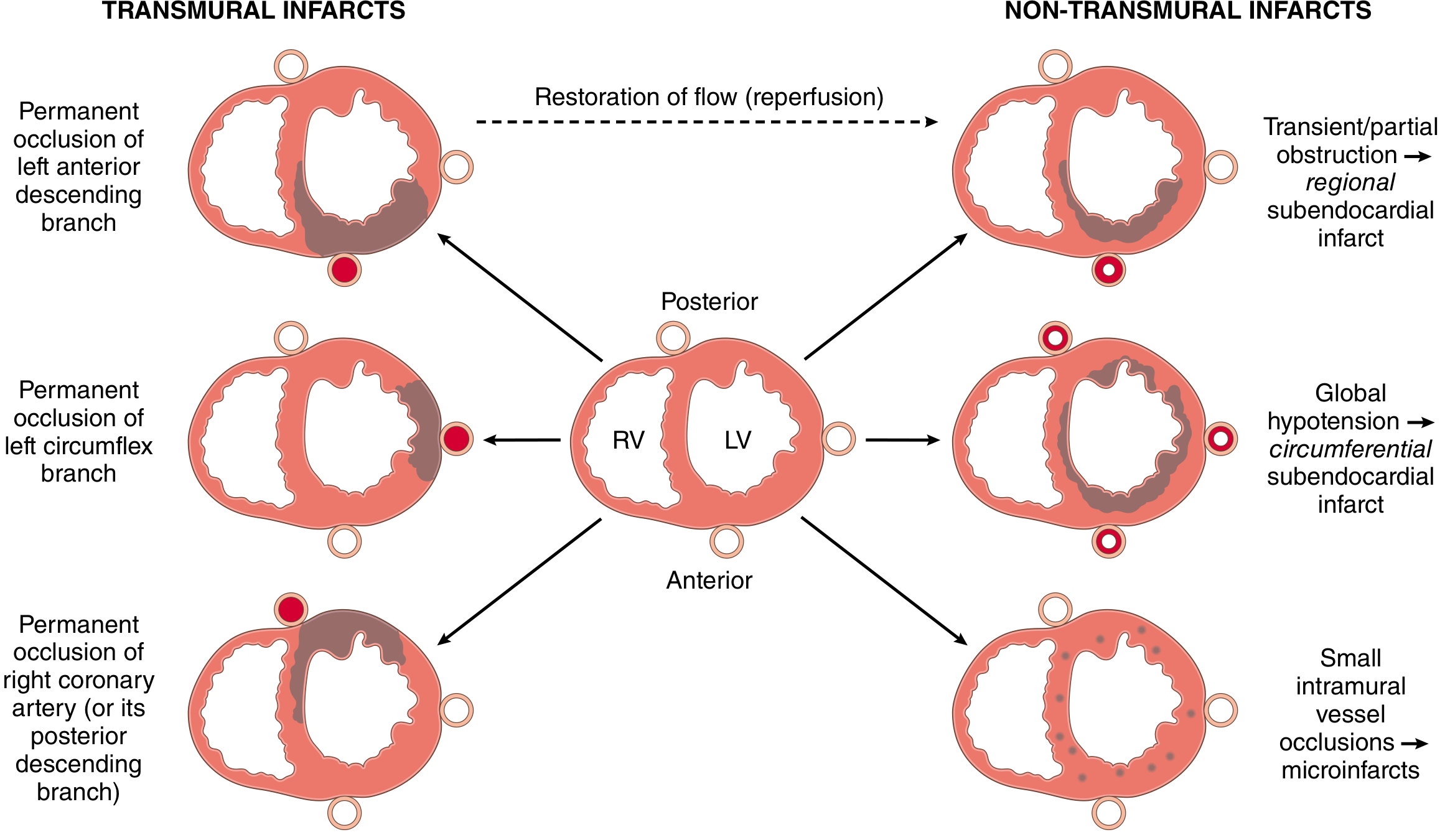
Fig. 12.11 from Robbins, Cotran & Kumar - Transmural vs. non-transmural infarct patterns based on type and location of coronary occlusion
4. Morphological Evolution (Gross & Microscopic)
| Time | Gross Changes | Microscopic Changes |
|---|---|---|
| 0-4 hours | None visible | Wavy fibers, early coagulation necrosis |
| 4-12 hours | Pale or mottled | Coagulation necrosis; edema; hemorrhage |
| 12-24 hours | Pallor; occasionally hyperemia | Neutrophil infiltration begins |
| 1-3 days | Pale/tan, soft | Heavy neutrophil infiltration, necrosis with nuclear pyknosis |
| 3-7 days | Yellow-tan, soft center; hyperemic border | Macrophage infiltration begins; removal of necrotic debris |
| 1-3 weeks | Yellow-tan, bordered by red hyperemic zone | Granulation tissue with fibroblasts and new vessels |
| 1-2 months | White fibrous scar forming | Dense collagen fibrosis |
| > 2 months | Dense white scar | Dense collagen scar |
- Robbins, Cotran & Kumar Pathologic Basis of Disease
5. Clinical Features
Symptoms
- Chest pain (most common): crushing/squeezing substernal pressure that may radiate to left arm, jaw, neck, or back. Typically lasting > 20-30 minutes and not relieved by rest or nitrates.
- Associated symptoms: diaphoresis, nausea/vomiting, dyspnea, lightheadedness, sense of impending doom.
- Silent MI (no pain): especially in diabetics (due to neuropathy) and the elderly - may present only with dyspnea, fatigue, or syncope.
ECG Changes
The three major electrocardiographic changes in acute MI reflect three membrane defects:
| Defect in Infarcted Cells | Current Flow | ECG Change in Leads over Infarct |
|---|---|---|
| Rapid repolarization (accelerated K+ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of K+) | Into infarct during diastole | TQ depression (manifest as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
-
Hyperacute phase: tall peaked T waves
-
Acute phase (minutes to hours): ST segment elevation in leads overlying infarction (STEMI); reciprocal ST depression in opposite leads
-
Days to weeks: T wave inversions develop; ST gradually normalizes
-
Weeks later: Q waves appear (electrically silent scar) - "pathological Q waves" (>40ms wide, >25% of QRS height)
-
Ganong's Review of Medical Physiology, p. 534
Biomarkers
| Marker | Rises | Peaks | Returns to Normal |
|---|---|---|---|
| Troponin I/T (high-sensitivity) | 3-4 hours | 24-48 hours | 7-14 days |
| CK-MB | 4-6 hours | 12-24 hours | 48-72 hours |
| Myoglobin | 1-2 hours | 4-6 hours | 24 hours |
High-sensitivity cardiac troponin (hsTn) is the gold standard biomarker. Elevated troponin levels reflect sarcolemmal disruption allowing intracellular proteins to escape into the circulation. The earliest detectable feature of myocyte necrosis is disruption of the sarcolemmal membrane.
6. Classification: STEMI vs. NSTEMI
| Feature | STEMI | NSTEMI/UA |
|---|---|---|
| Mechanism | Complete occlusion | Partial/transient occlusion |
| ECG | ST elevation, Q waves | ST depression, T-wave changes, or normal |
| Troponin | Elevated | Elevated (NSTEMI) or normal (UA) |
| Infarct pattern | Transmural | Subendocardial |
| Urgency | Immediate reperfusion | Risk-stratified management |
7. Management
Immediate (All ACS)
- Oxygen (if SpO2 < 90%)
- Aspirin 300 mg loading dose (antiplatelet, irreversible COX-1 inhibition)
- P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) for dual antiplatelet therapy (DAPT)
- Anticoagulation: heparin (UFH or LMWH)
- Nitrates for pain relief (avoid in RV infarction or hypotension)
- Morphine for refractory pain (used cautiously - may delay P2Y12 absorption)
- Beta-blockers (early oral, if no contraindications - reduces heart rate and oxygen demand)
STEMI - Reperfusion Therapy
The cornerstone of STEMI management is rapid reperfusion:
- Primary PCI (percutaneous coronary intervention): preferred if available within 90 minutes of first medical contact ("door-to-balloon time"). Angioplasty ± stenting directly opens the occluded artery.
- Fibrinolysis (thrombolysis): if PCI is not available within 120 minutes. Agents: alteplase, tenecteplase, streptokinase. Effective if given within 12 hours of symptom onset.
- The benefit of reperfusion is greatest when achieved quickly - this is the rationale for the "time is muscle" principle, since only severe ischemia lasting 20-40 minutes or longer causes irreversible necrosis.
NSTEMI/UA - Medical Management + Early Invasive Strategy
- Risk stratification using GRACE or TIMI score
- Early invasive angiography (within 24-48 hours) for high-risk patients
Long-term Secondary Prevention
- Aspirin lifelong
- P2Y12 inhibitor (DAPT for 12 months post-ACS, then reassessed)
- Statin (high-intensity, e.g., atorvastatin 40-80 mg)
- ACE inhibitor / ARB (reduces LV remodeling, especially if reduced EF)
- Beta-blocker (reduces mortality post-MI, especially with reduced EF)
- Aldosterone antagonist (eplerenone/spironolactone if EF < 40% with symptoms)
Recent Evidence Note: A 2025 meta-analysis (PMID 39298680) questions the routine use of beta-blockers for secondary prevention in post-MI patients without reduced ejection fraction or heart failure, suggesting the benefit may be less clear in this subgroup - a potential update to classic teaching.
8. Causes of Death
The most common causes of death following acute MI are:
- Decreased cardiac output - systolic dysfunction; the non-contracting infarcted zone paradoxically bulges outward ("systolic stretch"), worsening overall pump function. Cardiogenic shock occurs when >40% of LV mass is infarcted.
- Pulmonary edema - from LV failure causing blood to dam back into pulmonary circulation.
- Ventricular fibrillation - the most common cause of sudden cardiac death in the first few hours; due to electrical instability in peri-infarct zone.
- Cardiac rupture - most common 3-7 days post-MI when necrotic tissue is softest; can cause hemopericardium and tamponade.
- Guyton and Hall Textbook of Medical Physiology, p. 271
9. Complications
Early (hours to days)
- Arrhythmias: VF, VT, heart block (especially with inferior MI and RCA occlusion), AF
- Cardiogenic shock: poor prognosis; mortality 50-80% without intervention
- Acute mitral regurgitation: from papillary muscle rupture (typically posteromedial papillary, day 2-7)
- Free wall rupture: pericardial tamponade, rapid death
- Ventricular septal defect (VSD): septal rupture, leads to acute left-to-right shunt
- Right ventricular infarction (with inferior STEMI): hypotension, elevated JVP, clear lungs
Late (days to weeks)
- Dressler syndrome (post-MI pericarditis): autoimmune reaction, occurs 2-10 weeks post-MI
- Left ventricular aneurysm: dyskinetic wall segment; risk of mural thrombus and systemic embolism
- Heart failure / dilated cardiomyopathy: from extensive infarction and adverse LV remodeling
- Sudden cardiac death from recurrent arrhythmias
A 2024 meta-analysis (PMID 39378245) found that the risk of sudden cardiac death post-MI remains elevated across long-term follow-up, with pooled individual participant data from large cohorts.
Summary Diagram of Pathophysiology
Atherosclerotic plaque rupture
↓
Platelet aggregation + thrombus
↓
Coronary artery occlusion
↓
Ischemia → ATP depletion (seconds)
↓
Loss of contractility (< 2 min)
↓
Irreversible necrosis (20-40 min) → "Time is muscle"
↓
Coagulation necrosis → Scar formation (weeks)
↓
Complications (arrhythmias, pump failure, rupture, remodeling)
Key Sources:
- Robbins, Cotran & Kumar - Pathologic Basis of Disease, Ch. 12 (Pathogenesis, morphology, epidemiology)
- Guyton & Hall Textbook of Medical Physiology (Physiology, causes of death)
- Ganong's Review of Medical Physiology (ECG changes)
- Robbins & Kumar Basic Pathology (Overview)
- Beta-blockers post-MI meta-analysis, Eur J Prev Cardiol 2025 (Updated evidence on secondary prevention)
This is a shared conversation. Sign in to Orris to start your own chat.