DKA
Reading File
Finding Sources
Finding Sources
Searching PubMed
"diabetic ketoacidosis" management
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content. Let me compile the full DKA overview:
Diabetic Ketoacidosis (DKA)
Definition
DKA is defined by three core biochemical features (the "DKA triad"):
- Hyperglycemia - blood glucose typically >250 mg/dL (though euglycemic DKA at ≤300 mg/dL occurs in up to 18%, especially with SGLT2 inhibitor use)
- Ketonemia/Ketonuria - urine ketones ≥2+ or serum ketones ≥3.0 mmol/L
- Metabolic acidosis - arterial/venous pH <7.3, serum bicarbonate <18 mmol/L
Mild DKA: pH 7.20-7.30; Severe DKA: pH <7.00.
- Goldman-Cecil Medicine, p. 2483
Pathophysiology
The central defect is insulin deficiency + glucagon excess, creating a state that mimics starvation at the cellular level.
Metabolic consequences:
| Pathway | Effect |
|---|---|
| Lipolysis (hormone-sensitive lipase activated) | Free fatty acids (FFAs) released from adipose tissue |
| Hepatic beta-oxidation of FFAs | Ketone body production: β-hydroxybutyrate, acetoacetate, acetone |
| Proteolysis | Amino acids → liver → gluconeogenesis |
| Glucogenolysis | Further raises blood glucose |
| Osmotic diuresis | Profound dehydration + electrolyte loss |
Glucose in the renal tubules draws water, Na+, K+, Mg2+, Ca2+, phosphorus into urine. Combined with vomiting and poor intake, this produces the profound dehydration and electrolyte imbalances of DKA. - Rosen's Emergency Medicine, p. 2542
Why ketones accumulate:
Despite increased ketone production, peripheral tissues reduce their use of ketones (mimicking starvation physiology). The body's own protective mechanism thus backfires, amplifying ketoacidosis.
Average fluid/electrolyte deficits in severe DKA:
- Water: 70-120 mL/kg
- Sodium: 8-10 mEq/kg
- Potassium: 5-7 mEq/kg
- Phosphorus: ~3 mEq/kg
- Rosen's Emergency Medicine, Table 115.3
Precipitants
| Most Common | Other |
|---|---|
| Infections | Cerebrovascular accident |
| Inadequate insulin / non-adherence | Acute pulmonary embolism |
| New-onset diabetes | Pancreatitis |
| Acute coronary syndrome | Cushing syndrome, thyrotoxicosis |
| Unknown | Drugs: corticosteroids, clozapine, olanzapine, cocaine, SGLT2 inhibitors, thiazides |
- Goldman-Cecil Medicine, Table 210-11
Clinical Features
- Prodrome (hours to days): polyuria, polydipsia, weakness, lethargy, nausea, anorexia
- Pain: nonspecific upper abdominal pain that can mimic acute abdomen; paralytic ileus in severe cases
- Dehydration signs: dry skin and mucous membranes, decreased JVP, tachycardia, orthostatic hypotension
- Neurological: depressed mental status, frank coma in severe cases
- Respiratory: Kussmaul breathing - deep, rapid respirations as respiratory compensation for metabolic acidosis; fruity breath (acetone)
- Goldman-Cecil Medicine, p. 2484
Diagnosis & Lab Findings
| Test | Findings in DKA |
|---|---|
| Glucose | Usually >350 mg/dL; euglycemic DKA possible |
| pH (venous preferred) | <7.3 (venous correlates well with arterial, less invasive) |
| Bicarbonate | <18 mEq/L |
| Anion gap | Elevated (from acetoacetate + β-hydroxybutyrate + lactate) |
| Potassium | Initially normal or HIGH (K+ shifts out of cells in exchange for H+), but total body K+ is DEPLETED |
| Sodium | Often low (dilutional from osmotic water shift); correct for hyperglycemia |
| Ketones | Urine ≥2+ or serum β-hydroxybutyrate ≥3 mmol/L |
| WBC | Elevated even without infection (due to acidosis itself) |
| Amylase | Often elevated - usually non-pancreatic origin |
Key pitfall on ketone testing: Nitroprusside-based strips only detect acetoacetate. β-hydroxybutyrate (the predominant ketone in DKA) does NOT react - so dipstick ketones can be falsely low. Use serum β-hydroxybutyrate when available.
Corrected sodium: For every 100 mg/dL glucose above 100, add 1.6-2.4 mEq/L to measured Na+.
Winter's formula to check respiratory compensation:
Expected PaCO₂ = (1.5 × HCO₃⁻) + 8 ± 2
Delta gap (Delta AG - Delta HCO₃):
-
+6: coexisting metabolic alkalosis
-
<-6: coexisting hyperchloremic acidosis
-
Rosen's Emergency Medicine, pp. 2543-2544
Treatment
Treatment has four pillars: fluids, insulin, electrolytes, treat the precipitant.
1. Fluids
- Start 0.9% normal saline (even if osmolality is high - NS is still relatively hypotonic)
- Rate: 2-4 L in the first 2-4 hours in DKA
- Switch to 0.45% NaCl once corrected sodium is normal
- Add dextrose (D5 or D10) once glucose falls to 250 mg/dL to allow continued insulin infusion without causing hypoglycemia
2. Potassium (CRITICAL - must be addressed BEFORE insulin)
| Serum K+ | Action |
|---|---|
| <3.5 mEq/L | Hold insulin, replace K+ aggressively (20-40 mEq/hr IV) |
| 3.5-5.5 mEq/L | Give 20-40 mEq K+ per liter of IV fluid, start insulin |
| >5.5 mEq/L | Hold K+ replacement, start insulin, recheck frequently |
ECG can help if immediate potassium level unavailable (look for hyperkalemia or hypokalemia signs).
3. Insulin
- Fixed-rate IV insulin infusion: 0.1 units/kg/hour
- Do NOT give insulin bolus unless glucose >500 mg/dL and no IV access available
- If the patient was already on a background long-acting subcutaneous insulin, continue it alongside IV insulin
- Once glucose falls to 250 mg/dL AND ketones <1.0 mmol/L: reduce infusion to 0.05 units/kg/hour
- Continue IV insulin until the patient can eat AND ketoacidosis is resolved, then overlap with subcutaneous insulin (give subcut dose 1-2 hours before stopping IV)
4. Bicarbonate
- Generally NOT recommended unless severe acidosis (pH <6.9) because:
- Paradoxical CNS acidosis
- Worsens hypokalemia
- Delays ketone clearance
- If used: 100 mEq NaHCO₃ over 2 hours with 20 mEq KCl
5. Phosphate
-
Routine replacement not generally recommended
-
Consider if symptomatic (respiratory weakness, cardiac dysfunction) or phosphate <1.0 mg/dL
-
Goldman-Cecil Medicine, pp. 2484-2485; Rosen's Emergency Medicine
DKA vs HHS - Key Differences
| Feature | DKA | HHS |
|---|---|---|
| Glucose | 250-800 mg/dL | Often >600 mg/dL (can exceed 1000) |
| pH | <7.3 | >7.3 |
| Bicarb | <18 mEq/L | >18 mEq/L |
| Ketones | Significant | Absent or trace |
| Osmolality | Mildly elevated | >320 mOsmol/L |
| Age/Type | Usually T1DM, younger | Usually T2DM, elderly |
| Onset | Hours-days | Days-weeks |
| Treatment | Fluids + insulin from start | Fluids first; insulin when glucose stops falling |
Complications to Monitor
- Cerebral edema - most feared complication, especially in children; risk increased by rapid correction of osmolality
- Hypokalemia - life-threatening, especially after insulin starts
- Hypoglycemia - from aggressive insulin without dextrose
- ARDS - especially in older adults
- Thromboembolic events - from hemoconcentration
Recent Evidence Note
Two recent meta-analyses (2024-2026) are relevant to insulin delivery strategy in DKA:
- A 2024 systematic review (PMID: 39090718) compared subcutaneous insulin vs. continuous insulin infusion - subcutaneous regimens showed non-inferior outcomes in selected patients.
- A 2026 meta-analysis of RCTs (PMID: 41208563) examined early subcutaneous basal insulin added to IV insulin infusion - supports the practice of overlapping subcutaneous basal insulin early to reduce rebound DKA after infusion discontinuation.
These may update classic IV-only protocols, particularly in stable, mild-moderate DKA in adults.
Chart formate
Here is the DKA content in chart/table format:
DKA - Quick Reference Charts
1. DEFINITION & SEVERITY
| Parameter | Mild | Moderate | Severe |
|---|---|---|---|
| Glucose | >250 mg/dL | >250 mg/dL | >250 mg/dL |
| pH | 7.25 - 7.30 | 7.00 - 7.24 | <7.00 |
| Bicarbonate | 15-18 mEq/L | 10-14 mEq/L | <10 mEq/L |
| Ketones (urine) | 2+ | 2-3+ | 3+ |
| Anion gap | >10 | >12 | >12 |
| Mental status | Alert | Alert/drowsy | Stupor/coma |
2. PATHOPHYSIOLOGY AT A GLANCE
| Defect | Consequence | End Result |
|---|---|---|
| Insulin deficiency + glucagon excess | Lipolysis activated | FFAs → ketones |
| Hormone-sensitive lipase activated | β-hydroxybutyrate + acetoacetate produced | Anion gap metabolic acidosis |
| Proteolysis + gluconeogenesis | Hepatic glucose output ↑ | Hyperglycemia |
| Hyperglycemia exceeds renal threshold | Osmotic diuresis | Dehydration + electrolyte loss |
| Intracellular acidosis | K+ shifts out of cells | Pseudohyperkalemia; total body K+ depleted |
3. PRECIPITANTS
| Category | Examples |
|---|---|
| Infection | UTI, pneumonia, sepsis |
| Insulin issues | Non-adherence, missed doses, pump failure |
| New diagnosis | First presentation of T1DM |
| Cardiac | Acute MI, ACS |
| Drugs | Corticosteroids, SGLT2 inhibitors, clozapine, olanzapine, cocaine, thiazides |
| Endocrine | Cushing syndrome, thyrotoxicosis, acromegaly |
| Other | CVA, PE, pancreatitis, burns, alcohol |
4. CLINICAL FEATURES
| System | Symptoms/Signs |
|---|---|
| General | Weakness, lethargy, malaise |
| Metabolic | Polyuria, polydipsia, weight loss |
| GI | Nausea, vomiting, abdominal pain, ileus |
| Respiratory | Kussmaul breathing (deep, rapid), fruity breath |
| Cardiovascular | Tachycardia, orthostatic hypotension |
| Skin/Mucosa | Dry skin, dry mucous membranes |
| CNS | Depressed consciousness → coma |
| Eyes | Reduced JVP, sunken eyes |
5. LAB FINDINGS
| Test | Expected Finding | Key Pitfall |
|---|---|---|
| Glucose | >250 mg/dL (euglycemic DKA possible with SGLT2i) | Can be normal in euglycemic DKA |
| pH (venous) | <7.30 | Venous preferred - less invasive, correlates well |
| Bicarbonate | <18 mEq/L | Vomiting can mask severity (pseudo-normal) |
| Anion gap | Elevated (>12) | May be normal if aggressive NS given |
| Potassium | Normal or HIGH initially | Total body K+ is depleted - will crash with insulin |
| Sodium | Low (dilutional) | Correct for glucose: +1.6 mEq/L per 100 mg/dL ↑ glucose |
| Ketones (dipstick) | Positive | Misses β-hydroxybutyrate - use serum BHB |
| WBC | Elevated | Not always infection - acidosis itself raises WBC |
| Amylase | Elevated | Usually non-pancreatic - does NOT mean pancreatitis |
6. TREATMENT PROTOCOL
| Step | Action | Details |
|---|---|---|
| 1. Fluids | 0.9% Normal Saline | 2-4 L in first 2-4 hours |
| Switch fluid | 0.45% NaCl once corrected Na+ is normal | |
| Add dextrose | D5 or D10 when glucose reaches 250 mg/dL | |
| 2. Potassium (check BEFORE insulin) | K+ <3.5 mEq/L | Hold insulin - replace K+ 20-40 mEq/hr first |
| K+ 3.5-5.5 mEq/L | Add 20-40 mEq K+ per liter IV fluid, start insulin | |
| K+ >5.5 mEq/L | Hold K+ replacement, start insulin, recheck q2h | |
| 3. Insulin | IV infusion | 0.1 units/kg/hour fixed rate |
| Reduce rate | When glucose ≤250 AND ketones <1.0 mmol/L → 0.05 units/kg/hr | |
| Transition | Give subcutaneous dose 1-2 hrs before stopping IV | |
| 4. Bicarbonate | Generally avoid | Only if pH <6.9 - risk: CNS acidosis, hypokalemia |
| 5. Phosphate | Usually not needed | Replace only if PO₄ <1.0 mg/dL or symptomatic |
| 6. Precipitant | Identify and treat | Cultures, ECG, imaging as needed |
7. POTASSIUM MANAGEMENT FLOWCHART
Check serum K+ FIRST
|
─────┼─────────────────
| |
K+ <3.5 K+ ≥3.5
| |
Hold insulin Start insulin 0.1 u/kg/hr
Replace K+ aggressively |
Recheck K+ ─────┼─────────────────
Then start insulin | |
K+ 3.5-5.5 K+ >5.5
Add 40 mEq/L Hold K+ in fluids
to IV fluids Recheck q2h
8. DKA vs HHS
| Feature | DKA | HHS |
|---|---|---|
| Glucose | 250-800 mg/dL | >600 mg/dL (often >1000) |
| pH | <7.30 | >7.30 |
| Bicarbonate | <18 mEq/L | >18 mEq/L |
| Ketones | Significant | Absent/trace |
| Serum osmolality | Mildly elevated | >320 mOsmol/L |
| Onset | Hours-days | Days-weeks |
| Typical patient | T1DM, younger | T2DM, elderly |
| Initial insulin | Yes, from start | Fluids first; insulin when glucose stops falling |
| Mortality | ~1-5% | ~10-20% |
9. COMPLICATIONS
| Complication | Timing | Notes |
|---|---|---|
| Hypokalemia | During treatment | Most common; life-threatening arrhythmias |
| Hypoglycemia | During treatment | If glucose not monitored hourly |
| Cerebral edema | During treatment | Most feared in children; from rapid osmolality correction |
| ARDS | During/after treatment | Especially in elderly |
| Thromboembolism | During illness | Hemoconcentration promotes clotting |
| Rebound DKA | After stopping IV insulin | Prevent by overlapping subcutaneous insulin |
10. MONITORING FREQUENCY
| Parameter | Frequency |
|---|---|
| Blood glucose | Every 1 hour |
| Serum K+ | Every 2 hours (more frequent if <3.5) |
| Venous pH / HCO₃ | Every 2-4 hours |
| Serum ketones / BHB | Every 2-4 hours |
| Fluid balance | Every 1 hour |
| ECG | Continuous cardiac monitoring |
Pathophysiology of dka
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Here is a complete, detailed breakdown of DKA pathophysiology with the official textbook diagram:
Pathophysiology of DKA
Master Diagram (Tintinalli's Emergency Medicine)
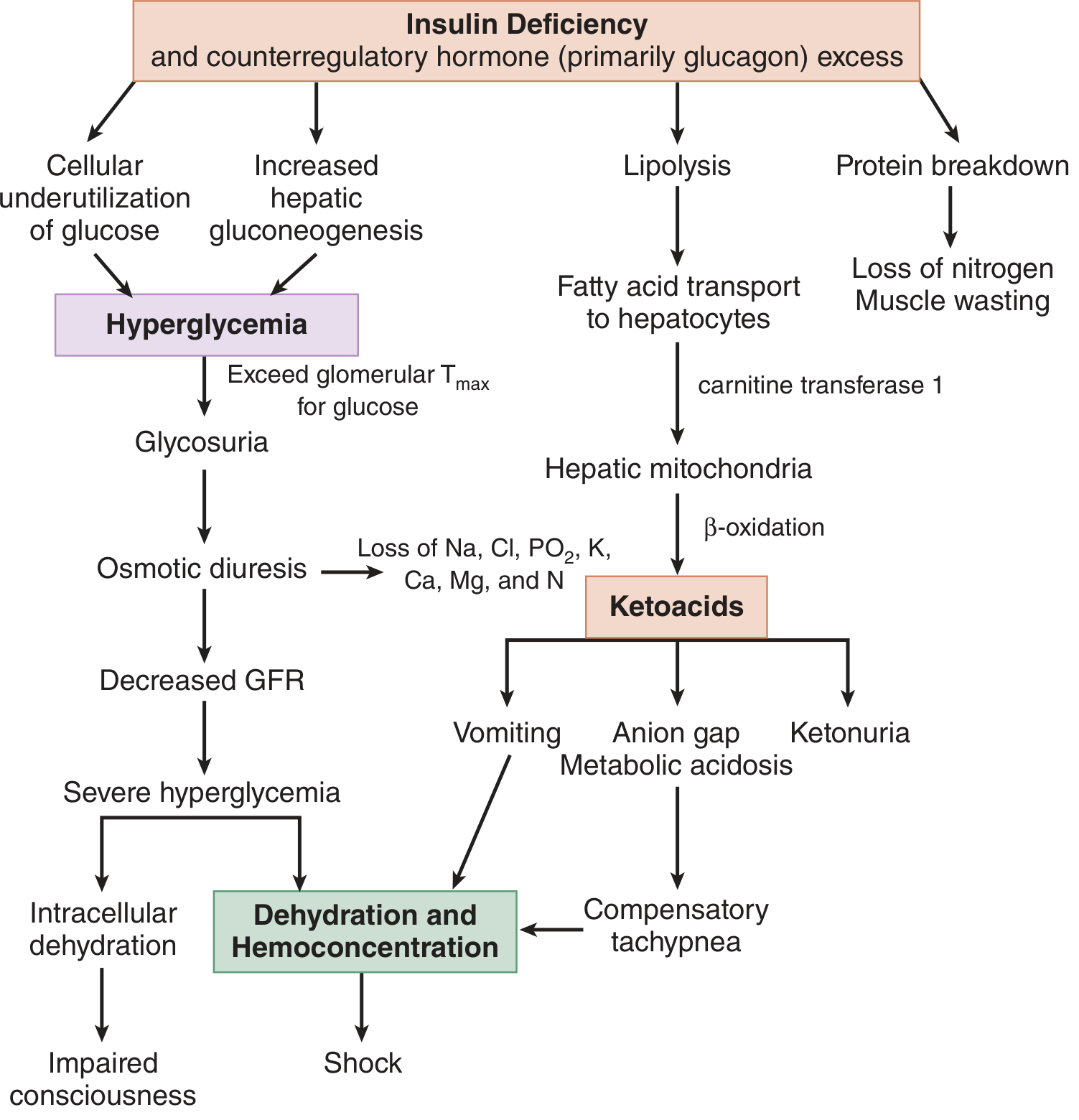
The Central Defect
Everything in DKA flows from one root cause:
Insulin deficiency + Counter-regulatory hormone excess (glucagon > catecholamines > cortisol > GH)
Insulin is the body's only anabolic hormone from the pancreas. Without it, the liver, adipose tissue, and skeletal muscle switch from storage mode to breakdown mode. The result is four simultaneous catastrophes: hyperglycemia, ketogenesis, dehydration, and acidosis.
Pathway 1 - HYPERGLYCEMIA
| Step | Mechanism |
|---|---|
| Insulin absent | Cells cannot take up glucose (GLUT-4 not activated in muscle/fat) |
| Glucagon excess | Activates glycogenolysis (breaks down liver glycogen) |
| Cortisol + catecholamines | Drive gluconeogenesis (amino acids + glycerol → glucose) |
| Proteolysis | Releases amino acids → liver → gluconeogenic precursors |
| Net result | Blood glucose rises far beyond renal T-max (~180 mg/dL) |
Key point: Even though glucose floods the bloodstream, cells are starving - "starvation in the midst of plenty." This drives further counter-regulatory hormone release, worsening the cycle.
Pathway 2 - KETOGENESIS
This is the most dangerous pathway and the one that distinguishes DKA from simple hyperglycemia.
Step-by-step:
1. Lipolysis activated
- Normally, insulin suppresses hormone-sensitive lipase (HSL) in adipose tissue
- With insulin absent, HSL is unrestrained
- Triglycerides are cleaved → free fatty acids (FFAs) + glycerol
- Glycerol → liver → gluconeogenesis (worsens hyperglycemia)
- FFAs → bound to albumin → transported to liver
2. FFAs enter hepatic mitochondria via carnitine shuttle
- Malonyl-CoA (which normally blocks the carnitine transferase-1 gate) is LOW in insulin deficiency
- FFAs freely enter mitochondria via carnitine acyltransferase-1 (CAT-1)
3. Beta-oxidation produces excess Acetyl-CoA
- FFAs are broken down to Acetyl-CoA
- Normally Acetyl-CoA enters the TCA cycle
- In insulin deficiency, TCA cycle is overwhelmed and oxaloacetate is depleted (diverted to gluconeogenesis)
- Excess Acetyl-CoA is shunted to ketogenesis
4. Three ketone bodies produced:
| Ketone | Notes |
|---|---|
| Acetoacetate | Detected by nitroprusside dipstick |
| β-hydroxybutyrate (BHB) | Predominant ketone in DKA; NOT detected by dipstick |
| Acetone | Volatile; causes fruity breath; weak dipstick reaction |
Ratio of BHB:acetoacetate is normally 1:1; in DKA rises to 3:1 or higher due to excess NADH from beta-oxidation shifting the equilibrium toward BHB.
5. Peripheral ketone utilization also falls
- Insulin normally promotes peripheral use of ketones as fuel
- Without insulin, brain, cardiac, and skeletal muscle reduce ketone uptake
- Combined with increased production → ketonemia accumulates rapidly
Pathway 3 - METABOLIC ACIDOSIS
| Source | Contribution to Acidosis |
|---|---|
| β-hydroxybutyrate | Major contributor to anion gap |
| Acetoacetate | Second major contributor |
| Lactate | Minor contributor (from hypoperfusion) |
| FFAs, phosphates | Minor contributors |
Anion gap = Na⁺ - (Cl⁻ + HCO₃⁻) - elevated because unmeasured anions (ketoacids) replace HCO₃⁻
Respiratory compensation: Acidemia stimulates the medullary respiratory center → Kussmaul breathing (deep, rapid) → CO₂ blown off → partially compensates pH
Additional complexity:
- Vomiting + loss of HCl → concurrent metabolic alkalosis can mask severity
- Aggressive NS resuscitation → hyperchloremic (non-AG) acidosis superimposed
- Ketonuria = loss of HCO₃⁻ equivalents (ketoanions excreted = potential bicarb lost); chloride retained in exchange
Pathway 4 - DEHYDRATION & ELECTROLYTE CHAOS
Osmotic Diuresis Cascade:
Hyperglycemia
↓
Glucose exceeds renal T-max → Glycosuria
↓
Osmotic diuresis (glucose in tubules drags water + electrolytes)
↓
Loss of: Na+, K+, Mg2+, PO4³⁻, Ca2+, Cl⁻ (5-10 L fluid lost)
↓
Volume depletion → ↓GFR → Less glucose excreted → Worse hyperglycemia
↓
Hemoconcentration → Further osmolality ↑
RAAS Activation:
- Volume depletion activates renin-angiotensin-aldosterone system
- Aldosterone causes further renal K+ wasting (on top of osmotic diuresis losses)
- This is why total body K+ is massively depleted even when serum K+ appears normal or high
Potassium Paradox (the most clinically dangerous electrolyte):
| Phase | Serum K+ | Total Body K+ | Why |
|---|---|---|---|
| Presentation | Normal or HIGH | DEPLETED | Acidosis drives K+ out of cells (H+/K+ exchange); insulin lack impairs cellular uptake |
| After insulin given | DROPS RAPIDLY | Already depleted | Insulin drives K+ back into cells; ongoing urinary losses |
| Risk | False reassurance → life-threatening hypokalemia |
Sodium Paradox:
- Measured Na+ is often LOW (dilutional - hyperglycemia draws water from cells into plasma)
- Correct: add 1.6 mEq/L to measured Na+ for every 100 mg/dL glucose >100
- True Na+ is actually higher than measured
Pathway 5 - VASCULAR & PROSTAGLANDIN EFFECTS
A less-known but important mechanism:
- As adipose tissue breaks down, prostaglandins I₂ and E₂ are released
- Both cause paradoxical vasodilation despite profound volume depletion
- This explains why peripheral vasodilation and flushing can occur even in shock states
- Also contributes to nausea, vomiting, and abdominal pain
Why Abdominal Pain Occurs in DKA
| Cause | Mechanism |
|---|---|
| Gastric dysmotility / ileus | Electrolyte imbalance (hypokalemia) + acidosis |
| Prostaglandin release | From lipolysis |
| Mesenteric vasodilation | Prostaglandins I₂ and E₂ |
| True pancreatitis | Can be precipitant OR consequence |
Important: Amylase is elevated in ~75% of DKA but is usually salivary origin, not pancreatic. Lipase is more specific.
Why Consciousness is Impaired
| Factor | Effect on CNS |
|---|---|
| Hyperosmolarity (from hyperglycemia + dehydration) | Most important determinant of mental status |
| Intracellular dehydration of neurons | Cellular shrinkage |
| Acidosis | Impairs neuronal function |
| Reduced cerebral perfusion | From hypovolemia/shock |
Complete Pathophysiology Summary Table
| Hormone/Defect | Target Organ | Effect | Clinical Result |
|---|---|---|---|
| ↓ Insulin | Liver | ↑ Glycogenolysis, ↑ gluconeogenesis | Hyperglycemia |
| ↓ Insulin | Adipose | HSL activation → lipolysis | FFAs → ketones |
| ↓ Insulin | Muscle | ↓ Glucose uptake, proteolysis | Amino acids → gluconeogenesis |
| ↑ Glucagon | Liver | ↑ Gluconeogenesis, ↑ ketogenesis, ↓ malonyl-CoA | Hyperglycemia + ketosis |
| ↑ Cortisol | Liver/muscle | ↑ Proteolysis, ↑ gluconeogenesis | Hyperglycemia |
| ↑ Catecholamines | Adipose | ↑ Lipolysis | More FFAs |
| ↑ Growth hormone | Muscle/adipose | Anti-insulin effects | Insulin resistance |
| Hyperglycemia | Kidney | Osmotic diuresis | Dehydration + electrolyte loss |
| ↓ Volume | RAAS | Aldosterone ↑ | Further K+ wasting |
| Acidosis | All cells | H+/K+ exchange | Pseudohyperkalemia |
| ↓ Insulin + ↓ K+ | Muscle/heart | Reduced K+ uptake | Hypokalemia after treatment |
| Ketoacids | Buffering system | HCO₃⁻ consumed | Anion gap metabolic acidosis |
| Low pH | Medulla | Respiratory drive ↑ | Kussmaul breathing |
- Tintinalli's Emergency Medicine, p. 1475-1476; Rosen's Emergency Medicine, p. 2542
This is a shared conversation. Sign in to Orris to start your own chat.