Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything I need for a comprehensive answer. Here is the full explanation:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Triggers
ARDS is an acute inflammatory response of the lung characterized by noncardiogenic pulmonary edema and refractory hypoxemia. The Berlin definition requires:
- Bilateral infiltrates on chest radiograph/CT not fully explained by effusions, collapse, or nodules
- PaO2/FiO2 ratio <300 mmHg (mild <300, moderate <200, severe <100)
- Onset within 7 days of a known clinical insult
- Respiratory failure not fully explained by heart failure or fluid overload
Common inciting triggers include sepsis (the leading cause), pneumonia, trauma, aspiration, blood product transfusion (TRALI), pancreatitis, and burns. Insults may be direct (pulmonary) or indirect (extrapulmonary).
Core Pathophysiological Concept
Unlike cardiogenic pulmonary edema - which results from elevated left-sided hydrostatic pressures - ARDS produces exudative pulmonary edema. The alveolar-capillary barrier develops increased permeability, allowing protein-rich fluid to flood the airspaces. This is not a pressure-driven process; it is an inflammatory one.
Three Sequential Phases
The pathological features of ARDS have classically been described as three overlapping and sequential stages:
Phase 1: Exudative Phase (Days 1-7)
This is the initial and defining phase. Key events:
-
Alveolar-capillary barrier disruption - Damage to both the pulmonary microvascular endothelium and alveolar epithelium increases barrier permeability. Loss of the epithelium disrupts barrier integrity and also prevents alveolar fluid clearance. Loss of endothelial barrier integrity is both necessary and sufficient for the development of ARDS.
-
Neutrophil sequestration - One of the earliest manifestations of ARDS (even before hypoxemia) is transient leukopenia due to neutrophil sequestration in the lung microvasculature. Because pulmonary capillaries are narrower than neutrophils, they must deform to pass through. Activated neutrophils become "stiff" via actin cytoskeletal changes and cannot negotiate these narrow segments, causing them to lodge in capillaries. They then migrate into the interstitium and alveoli, even without the usual adhesion molecules.
-
Neutrophil-mediated tissue injury - Sequestered neutrophils release a battery of cytotoxic compounds:
- Reactive oxygen species (ROS) - oxidative damage to membranes
- Neutrophil elastase (NE) - degrades epithelial and endothelial cadherins (adherens junction proteins), predisposing to alveolar flooding; also degrades surfactant protein A
- Metalloproteinases - further extracellular matrix degradation
- Cytokines: TNF-α, IL-1β, IL-8 - amplify inflammatory recruitment
- Neutrophil extracellular traps (NETs) - chromatin-based webs that trap but also damage surrounding tissue
-
Cytokine signaling cascade - Activated macrophages release TNF-α, IL-1β, IL-6, and IL-8. IL-8 is a potent neutrophil chemoattractant. p38 MAP kinase (activated by LPS) stimulates TNF-α and macrophage inflammatory protein-2, further driving neutrophil chemotaxis. This creates a positive-feedback inflammatory loop.
-
Alveolar flooding - Protein-rich fluid, cellular debris, and fibrin pour into alveolar spaces, forming hyaline membranes - a hallmark finding on histology (termed diffuse alveolar damage, or DAD).
-
Surfactant dysfunction - The proportion of large (active) to small (inactive) surfactant aggregates is diminished by decreased production and increased conversion. Leaked plasma proteins interfere with surfactant function. Neutrophil elastase degrades surfactant protein A. The net result is loss of alveolar surface tension reduction, promoting collapse.
-
Profound hypoxemia - Alveolar flooding causes ventilation-perfusion (V/Q) mismatch and right-to-left intrapulmonary shunting. Flooded or collapsed alveoli are perfused but not ventilated.
-
Decreased compliance - Fluid-filled and collapsed alveoli make the lung stiff; the functional residual capacity drops.
-
Pulmonary hypertension - Caused by hypoxic vasoconstriction, intravascular fibrin deposition, and compression of blood vessels by positive-pressure ventilation.
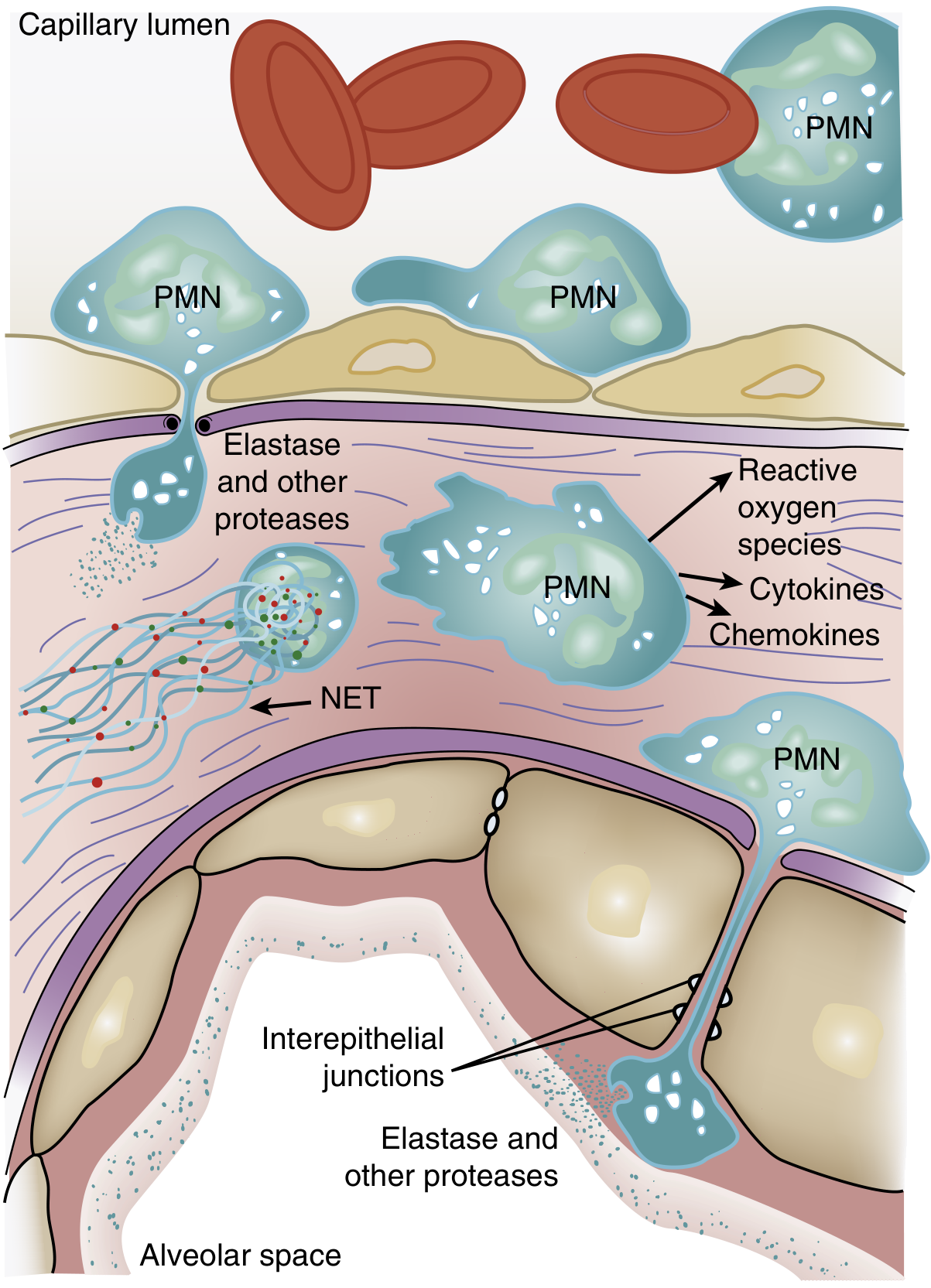
Phase 2: Proliferative Phase (Days 7-21)
In patients who survive the initial injury:
- Hyaline membranes begin to reorganize
- Type II pneumocyte hyperplasia - type II cells proliferate to replace destroyed type I pneumocytes and attempt to restore epithelial integrity
- Alveolar septal thickening develops
- Neutrophil numbers decrease; macrophages and lymphocytes increase
- Fibroblast proliferation begins in the interstitium and alveolar spaces
- In situ thrombi may appear in small pulmonary arteries
- Some resolution of pulmonary edema begins
Importantly, fibroproliferation may begin simultaneously with, not after, the inflammatory phase - elevated levels of N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid within 24 hours of disease onset.
Phase 3: Fibrotic Phase (>2 weeks, in a subset)
- Occurs in patients with persistent or severe ARDS
- Alveolar septal thickening from organizing fibrosis
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- BAL fluid from ARDS patients stimulates cultured fibroblasts to proliferate in vitro - suggesting active profibrotic signaling
Alveolar Fluid Clearance - Why It Fails
Normally, alveolar fluid is reabsorbed by vectorial sodium and chloride transport across type I and II alveolar epithelial cells, creating an osmotic gradient. In ARDS, this process is severely impaired because:
- Apoptosis and necrosis of the alveolar epithelium
- Defects in transcellular ion transport induced by proinflammatory cytokines, oxidants, and hypoxia
- Clotting of extravasated plasma proteins within airspaces slows protein removal (protein clearance is 1-2%/hour vs. fluid clearance at 10-20%/hour, leading to progressive alveolar protein accumulation)
Ventilator-Induced Lung Injury (VILI) and Biotrauma
A critical secondary mechanism: mechanical ventilation itself can worsen ARDS. High tidal volumes and inflation pressures (volutrauma/barotrauma) cause:
- Further endothelial and epithelial injury
- Enhanced neutrophil margination
- Increased release of proinflammatory cytokines from alveolar macrophages and epithelium (biotrauma)
- These mediators can spill into the systemic circulation, contributing to multi-organ dysfunction syndrome (MODS)
Additionally, high respiratory drive in spontaneously breathing patients can generate large negative intrathoracic pressures, causing patient self-inflicted lung injury (P-SILI).
Pathological Correlate: Diffuse Alveolar Damage (DAD)
DAD is considered the histological hallmark of ARDS, characterized by:
- Intra-alveolar hyaline membrane formation
- Interstitial and intra-alveolar edema
- Acute inflammation (neutrophil infiltration)
- Epithelial cell necrosis (temporal uniformity - suggesting a single acute insult)
- Later: type II pneumocyte hyperplasia, fibroblast proliferation, collagen deposition
However, DAD is only found in approximately 50% of ARDS patients on biopsy/autopsy. Patients with confirmed DAD tend to be younger, more severely ill, with poorer compliance and higher mortality.
Summary Cascade
Inciting insult (sepsis, pneumonia, trauma, etc.)
↓
Activation of alveolar macrophages + systemic inflammatory response
↓
Release of TNF-α, IL-1β, IL-6, IL-8, complement, platelet-activating factor
↓
Neutrophil sequestration in pulmonary capillaries
↓
Neutrophil transmigration → release of ROS, elastase, proteases, NETs
↓
Damage to alveolar epithelium + microvascular endothelium
↓
Increased alveolar-capillary permeability
↓
Protein-rich exudate floods alveoli → hyaline membrane formation
↓
Surfactant dysfunction → alveolar collapse
↓
V/Q mismatch + intrapulmonary shunt → refractory hypoxemia
↓
Decreased lung compliance + pulmonary hypertension
↓
[Ongoing] → proliferative → fibrotic phase
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine (Chapter 134: ARDS - Etiology, Pathogenesis, Pathology)
- Goldman-Cecil Medicine (Chapter 90/91: ARDS, Mechanical Ventilation)
- Fishman's Pulmonary Diseases and Disorders (Chapter 140: Resolution of Acute Lung Injury)
- Sabiston Textbook of Surgery (Chapter: Critical Care / ARDS)
This is a shared conversation. Sign in to Orris to start your own chat.