Dermatomyositis ILD full details for md exams 15marks answer from fishman and murray and ats and ers guidelines just from these only
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed for a comprehensive 15-mark MD exam answer. Let me compile this into a structured, detailed response.
Dermatomyositis - Interstitial Lung Disease (ILD)
15-Mark MD Examination Answer
Sources: Fishman's Pulmonary Diseases and Disorders (Chapter 58) | Murray & Nadel's Textbook of Respiratory Medicine (Chapter 92)
1. INTRODUCTION AND OVERVIEW
Dermatomyositis (DM) is a systemic autoimmune inflammatory myopathy distinguished from polymyositis (PM) by the presence of prominent skin involvement - the heliotropic rash (violaceous/purplish periorbital discoloration) and Gottron papules (erythematous scaly eruptions over the extensor surfaces of the finger joints). The disease belongs to the broader category of idiopathic inflammatory myopathies (IIMs).
Pulmonary complications are common and important causes of morbidity and mortality in DM. Pulmonary involvement has been reported in up to 40-45% of cases. In contrast to other connective tissue diseases (CTDs), in PM/DM, primary involvement of the airways and pleura does not routinely occur - the predominant pulmonary manifestation is parenchymal ILD.
- Fishman's Pulmonary Diseases and Disorders, p. 1031
- Murray & Nadel's Textbook of Respiratory Medicine, p. 2098
2. EPIDEMIOLOGY
-
Prevalence of ILD in PM/DM: 5% to 30% in most Western series; approaches 40-80% in Japanese populations in some series
-
When screened with HRCT, incidence approaches 78% within 3 years of diagnosis (Fishman's)
-
In larger series using BAL and HRCT for screening, documented incidence of diffuse lung disease can be up to 64% (Murray & Nadel)
-
ILD in DM is 3 to 5 times more common in women than men
-
Most commonly presents in the fifth decade
-
Bimodal age distribution of the underlying myopathies: childhood peak and fourth to fifth decade peak
-
HLA associations: HLA-B8/DR3, HLA-B14, HLA-B40 in DM; presence of diffuse lung disease has been associated with two specific HLA haplotypes
-
Inflammatory myopathies are relatively rare: 2 to 10 per 100,000 population, female-to-male ratio 2.5:1
-
Fishman's, p. 1031; Murray & Nadel, p. 2099
3. DIAGNOSTIC CRITERIA (Bohan and Peter Criteria)
| Criterion | Feature |
|---|---|
| 1 | Symmetrical proximal muscle weakness |
| 2 | Muscle biopsy specimen showing myositis |
| 3 | Elevation of serum skeletal muscle enzymes |
| 4 | Characteristic electromyographic pattern of myositis |
| 5 | Typical rash of DM (heliotrope rash / Gottron papules) |
For DM: Rash + any 3 of first 4 = definite; rash + 2 of first 4 = probable; rash + 1 of first 4 = possible.
Lung Manifestations listed by Murray & Nadel (Table 92.5):
- Interstitial pulmonary fibrosis
- Acute pneumonitis (with diffuse alveolar damage)
- Organizing pneumonia
- Aspiration pneumonia
- Pulmonary vasculitis and alveolar hemorrhage
- Respiratory muscle weakness
4. PULMONARY MANIFESTATIONS - FULL SPECTRUM
4.1 Interstitial Lung Disease (ILD) - The Most Important
Histological Patterns (in order of frequency):
| Pattern | Notes |
|---|---|
| NSIP (Nonspecific Interstitial Pneumonia) | Most common overall; 82% in biopsied patients in one large series (Douglas et al.); more cellular, more responsive to treatment |
| OP (Organizing Pneumonia) | Often admixed with NSIP; major component early; corticosteroid-responsive |
| UIP (Usual Interstitial Pneumonia) | Previously thought predominant; less common with revised classifications |
| DAD (Diffuse Alveolar Damage) | Acute presentation; associated with poorer prognosis; often refractory to treatment |
| DAH (Diffuse Alveolar Hemorrhage) | Due to pulmonary capillaritis; less common |
Key point from Fishman's: While UIP was previously reported as predominant, NSIP with organizing pneumonia now appears most common based on the revised IIP classification system.
Temporal relationship: ILD may precede, appear simultaneously with, or follow the muscle/skin manifestations. In fact, ILD may precede joint and muscle disease by several months to years.
Important negative correlations (Fishman's): There is NO relationship between ILD and:
-
Extent of muscle or skin disease
-
Level of creatine phosphokinase (CPK) elevation
-
Presence of serum rheumatoid factor or antinuclear antibodies
-
Fishman's, p. 1031; Murray & Nadel, p. 2099
4.2 Aspiration Pneumonia
- Occurs in 10-20% of patients with PM/DM (Murray & Nadel: up to 20%)
- Mechanism: Inflammatory myositis affecting the striated muscle of the hypopharynx and upper one-third of the esophagus → loss of normal swallowing function → failure to protect the airway
- Almost half of these patients complain of dysphagia
- More likely in patients with extensive skin or muscle involvement
4.3 Respiratory Muscle Dysfunction
- Hypercapnic respiratory failure requiring assisted ventilation occurs in approximately 5% of patients (Fishman's) or up to 25% (Murray & Nadel) due to extensive myositis involving respiratory muscles and diaphragm
- With less extensive involvement: reduction in cough generation → hypostatic pneumonia, atelectasis from mucus plugging
- Weakness causes a restrictive physiologic defect with tachypnea and dyspnea despite normal diffusing capacity and normoxia
- Best demonstrated by measurement of maximal pressures generated during both phases of the respiratory cycle - useful for monitoring disease course and treatment response
- Bilateral diaphragmatic paralysis has been reported
4.4 Pulmonary Arterial Hypertension
- WHO Group I PAH has been reported but is uncommon
- Most often in cases with suspected crossover with scleroderma
- Patients present with dyspnea and fatigue, normal chest radiographs except for pulmonary arterial enlargement and isolated reduction in DLCO
5. AUTOANTIBODIES AND THE ANTISYNTHETASE SYNDROME
This is the most examinable section and directly links serology to pulmonary manifestations.
Anti-Jo-1 (anti-histidyl tRNA synthetase):
- Present in 25% of all patients with PM/DM
- Present in 50-100% of patients with PM/DM + ILD
- Present in only 5-13% of patients without ILD
- Most common of the antisynthetase antibodies
Other antisynthetase antibodies: PL-12, PL-7, EJ, OJ, Ku - all associated with ILD
Anti-MDA5 (Melanoma Differentiation-Associated gene 5):
- Associated with rapidly progressive ILD
- Often amyopathic DM (rash without significant muscle involvement)
- Particularly relevant given its association with acute, treatment-refractory ILD
The Antisynthetase Syndrome (Murray & Nadel Table 92.6):
A patient must have a positive anti-tRNA antibody PLUS one or more of:
- Myositis (by Bohan and Peter criteria)
- ILD (by ATS criteria)
- Arthritis
- Unexplained, persistent fever
- Raynaud phenomenon
- Distal digital fissuring ("mechanic's hands")
Key clinical pearl from Murray & Nadel: Low creatine kinase levels have been associated with more rapidly progressive diffuse lung disease - counterintuitive but important. Several studies report NO association between CK levels and respiratory disease severity.
- Fishman's, p. 1031; Murray & Nadel, p. 2100
6. CLINICAL FEATURES OF DM-ILD
Symptoms:
- Dyspnea and nonproductive cough are the most common presenting symptoms
- Breathlessness on exertion without wheeze
- If myopathy is severe: orthopnea may be striking
- Hemoptysis if capillaritis is present
- Pleural disease is uncommon
- No correlation between severity of pulmonary involvement and systemic musculoskeletal manifestations
- Rarely, patients present acutely with or progress to acute respiratory failure
Timing: Pulmonary involvement can develop before the systemic disease, at any time during the disease course, or after established muscle/skin disease.
7. IMAGING
Chest Radiograph
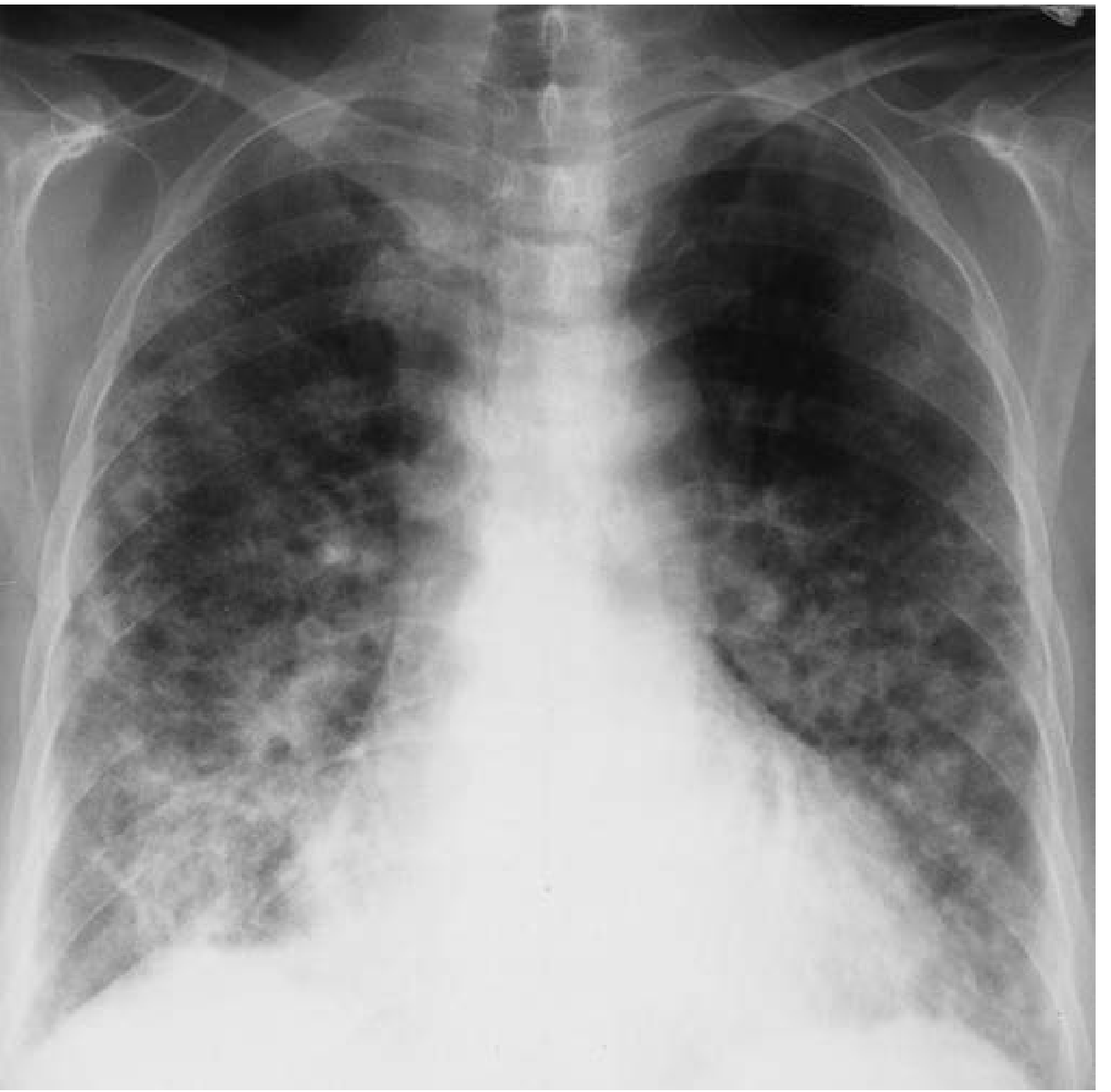
Fig: Organizing pneumonia in PM/DM: diffuse patchy alveolar infiltrates (Fishman's, Fig. 58-20)
- Chronic ILD: bibasilar reticulonodular infiltrates; with progression, reduced lung volume, honeycomb lung
- Acute pneumonitis (DAD): diffuse ground-glass opacification
- Alveolar hemorrhage: areas of consolidation
- With disease progression: reduction of lung volumes, development of radiographic honeycomb lung and pulmonary hypertension
HRCT
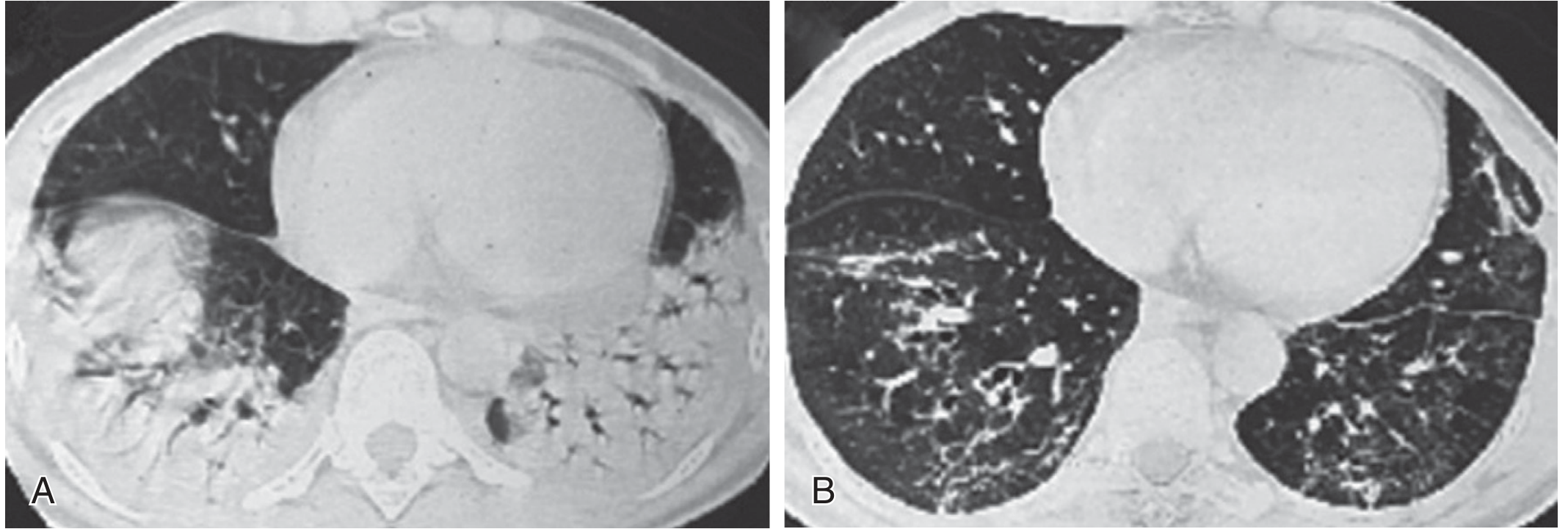
Fig: (A) Dense posterior consolidation of organizing pneumonia in PM; (B) Post-treatment: regression of consolidation with residual linear opacities and traction bronchiectasis (Murray & Nadel, Fig. 92.5)
HRCT is more sensitive and specific than chest radiography. CT findings include:
- Thickened interlobular septa
- Linear opacities
- Ground-glass opacification (prominent early feature)
- Patchy consolidation
- Peripheral reticular pattern - combination of consolidation with peripheral reticular pattern is highly characteristic
- Traction bronchiectasis and honeycombing (less common, seen with fibrotic disease)
- The consolidation may evolve into a reticular pattern over time
- HRCT screening increases documented ILD incidence to approximately 78% within 3 years
NSIP on HRCT: Bilateral ground-glass opacity with or without reticulation, basal predominant, subpleural sparing (in some cases)
OP on HRCT: Dense posterior lower-lobe consolidation, most prominent posteriorly
- Fishman's, p. 1031; Murray & Nadel, p. 2099
8. PULMONARY FUNCTION TESTS
- Restrictive ventilatory defect: Reduced TLC, FVC, FEV1 with preserved FEV1/FVC ratio
- Reduced DLCO - severe reduction associated with increased mortality
- With recent hemorrhage or marked myopathy: disproportionate preservation of DLCO (because hemorrhage artificially elevates CO uptake)
- Acute hemorrhage can present in context of previous chronic disease; DLCO may be normal or subnormal but elevated from a previously lower level
- Respiratory muscle weakness alone can cause restrictive defect with normal diffusing capacity and normoxia (distinguishes it from parenchymal ILD)
9. BRONCHOALVEOLAR LAVAGE (BAL)
- BAL lymphocytosis AND neutrophilia have been described in PM/DM-associated ILD
- Neutrophilia on BAL is associated with clinical deterioration (Murray & Nadel)
- BAL helps exclude infectious etiologies before initiating immunosuppression
10. LABORATORY INVESTIGATIONS
| Investigation | Finding |
|---|---|
| Serum CK | Elevated in active myositis; NOT correlated with ILD severity |
| Anti-Jo-1 | Positive in 50-100% of DM/ILD cases; indicates antisynthetase syndrome |
| Anti-MDA5 | Rapidly progressive ILD, often amyopathic DM |
| Anti-PL-12, PL-7, EJ, OJ | Other antisynthetase antibodies |
| ANA | May be positive |
| RF | Present in some; NOT related to ILD |
| BAL | Lymphocytosis ± neutrophilia |
Note from Murray & Nadel: Screening for occult malignancy is recommended in DM/PM (mammography, abdominal CT, pelvic ultrasonography, tumor markers, PET in some settings) because DM is a well-recognized paraneoplastic phenomenon.
11. ACUTE vs. SUBACUTE vs. CHRONIC ILD PRESENTATIONS
| Presentation | Underlying Lesion | Prognosis | Response to Treatment |
|---|---|---|---|
| Chronic | NSIP (cellular or fibrotic) | Better for cellular NSIP | Good - steroid responsive |
| Subacute | OP ± NSIP | Intermediate | Good - corticosteroid responsive |
| Acute/Rapidly Progressive | DAD | Poor | Usually refractory - "recovery is unusual" |
| With hemoptysis | Pulmonary capillaritis/DAH | Intermediate | Corticosteroids + cyclophosphamide |
- Fishman's, p. 1031-1032
12. TREATMENT
First-Line
Corticosteroids are the preferred initial therapy:
- Oral prednisolone 0.75-1 mg/kg/day for stable/subacute disease
- IV corticosteroids required for severe or rapidly progressive disease
Second-Line (Steroid-Resistant or Steroid-Sparing)
The following have been used with efficacy per both textbooks:
| Agent | Notes |
|---|---|
| Cyclophosphamide | For DAH (with corticosteroids), severe/refractory disease |
| Cyclosporin A | Calcineurin inhibitor; used in steroid-resistant cases |
| Tacrolimus | Calcineurin inhibitor; multiple reports of efficacy in PM/DM-ILD |
| Azathioprine | Steroid-sparing agent |
| Mycophenolate mofetil (MMF) | Steroid-sparing; used with increasing frequency |
| IV Immunoglobulin (IVIG) | Considered in refractory cases |
| Rituximab | Promising for antisynthetase syndrome; retrospective studies show CTD-ILD stabilization; myositis-associated ILD may represent the subgroup with greatest response to rituximab |
Key points:
-
Response to treatment depends on underlying histology - more cellular disease is more responsive (Fishman's)
-
NSIP/OP patterns: corticosteroid responsive
-
DAD pattern: poor response; "recovery is unusual despite aggressive anti-inflammatory and immunosuppressive therapy" (Fishman's)
-
DAH due to capillaritis: immunosuppression with corticosteroids + cyclophosphamide is utilized and has been efficacious
-
In refractory cases: IVIG should be considered; rituximab is promising, especially for antisynthetase syndrome including life-threatening disease refractory to other immunomodulatory therapies
-
Fishman's, p. 1032; Murray & Nadel, p. 2100-2101
13. PROGNOSIS
- ILD is a serious complication associated with increased mortality
- DM/PM are the most frequent cause of death from pulmonary complications (~45% of patients affected)
- Severe reduction in DLCO is associated with increased mortality
- Clinical course is heterogeneous and depends on histologic pattern
- Rapidly progressive ILD (especially with anti-MDA5 antibodies or DAD pattern) carries the worst prognosis
- BAL neutrophilia is associated with clinical deterioration
- DM/PM are well-recognized paraneoplastic phenomena - underlying malignancy must be excluded
14. INTERSTITIAL PNEUMONIA WITH AUTOIMMUNE FEATURES (IPAF)
A proportion of patients with IIP have autoimmune signs/symptoms but fail to meet established CTD criteria. The ERS/ATS Task Force established the IPAF concept with three domains:
- Clinical domain - specific extrathoracic features
- Serologic domain - specific autoantibodies (including antisynthetase antibodies)
- Morphologic domain - radiographic, pathologic, and physiologic features
In most CTDs other than RA, NSIP is the most common radiographic and pathologic pattern. UIP pattern is more common in IPAF (65%) compared to NSIP. Acute exacerbations may be less frequent in IPAF and more responsive to anti-inflammatory therapy.
- Fishman's, p. 1033-1034
15. SUMMARY TABLE FOR QUICK REVISION
| Feature | Key Detail |
|---|---|
| ILD prevalence | 5-30% (West); 40-80% (Japan); up to 78% on HRCT screening |
| Predominant histology | NSIP > OP > UIP > DAD |
| Key autoantibody | Anti-Jo-1 (50-100% of DM+ILD cases) |
| Rapidly progressive ILD marker | Anti-MDA5 |
| Antisynthetase syndrome | Anti-tRNA Ab + myositis/ILD/arthritis/Raynaud/mechanic's hands/fever |
| ILD vs muscle disease correlation | None - ILD may precede muscle disease |
| CK and ILD | No correlation; low CK = more rapidly progressive ILD |
| Imaging hallmark | Peripheral reticular + consolidation (HRCT); bibasilar reticulonodular (CXR) |
| BAL | Lymphocytosis + neutrophilia; neutrophilia = clinical deterioration |
| PFT | Restrictive + reduced DLCO; severe DLCO reduction = increased mortality |
| First-line treatment | Prednisolone 0.75-1 mg/kg/day (oral) or IV for severe/RP-ILD |
| Best response to treatment | Cellular NSIP, OP patterns |
| Worst prognosis pattern | DAD - "recovery is unusual" |
| DAH treatment | Corticosteroids + cyclophosphamide |
| Refractory ILD | Rituximab, IVIG |
| Malignancy screening | Mandatory - DM is a paraneoplastic phenomenon |
Sources:
- Fishman's Pulmonary Diseases and Disorders, 2-Volume Set, Chapter 58: Pulmonary Manifestations of Collagen Vascular Diseases (pp. 1029-1034)
- Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set, Chapter 92: Pulmonary Manifestations of Connective Tissue Diseases (pp. 2098-2101, Table 92.5, Table 92.6)
This is a shared conversation. Sign in to Orris to start your own chat.