General pharmacy chapter for bds students
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive content from the textbooks. Here is the complete General Pharmacy chapter for BDS students:
General Pharmacy - BDS Students
Sources: Lippincott Illustrated Reviews: Pharmacology | Katzung's Basic & Clinical Pharmacology, 16e | Goodman & Gilman's The Pharmacological Basis of Therapeutics
1. Definition of Pharmacology
Pharmacology is the study of substances that interact with living systems through chemical processes, by binding to regulatory molecules and activating or inhibiting normal body processes.
Two core branches:
- Pharmacokinetics - what the body does to the drug (absorption, distribution, metabolism, elimination - ADME)
- Pharmacodynamics - what the drug does to the body (mechanism of action, receptor interactions)
- Toxicology - the branch dealing with undesirable effects of chemicals on living systems
"Pharmacokinetics refers to what the body does to a drug, whereas pharmacodynamics describes what the drug does to the body." - Lippincott Illustrated Reviews: Pharmacology
2. Drug Sources and Properties
Sources of Drugs
| Source | Examples |
|---|---|
| Plant | Morphine (opium poppy), Atropine (Atropa belladonna), Digoxin (foxglove) |
| Animal | Insulin (pancreas), Heparin |
| Mineral | Lithium, Iron salts |
| Synthetic | Most modern drugs |
| Microbiological | Penicillin, Streptomycin |
| Recombinant DNA | Erythropoietin, Growth hormone |
Drug Size
Most drugs have molecular weights (MW) between 100 and 1000. Drugs with MW > 1000 do not diffuse readily between body compartments and must be administered directly into their target compartment (e.g., alteplase given IV for clot dissolution).
Drug-Receptor Bonds
Three major types:
- Covalent bonds - very strong, often irreversible (e.g., aspirin irreversibly acetylates cyclooxygenase - this is why aspirin's antiplatelet effect lasts days)
- Electrostatic bonds - most common; includes ionic bonds, hydrogen bonds, and van der Waals forces
- Hydrophobic bonds - weak; important for lipid-soluble drugs at cell membranes
Drugs binding through weak bonds are generally more selective because they require a very precise fit to their receptor.
3. Routes of Drug Administration
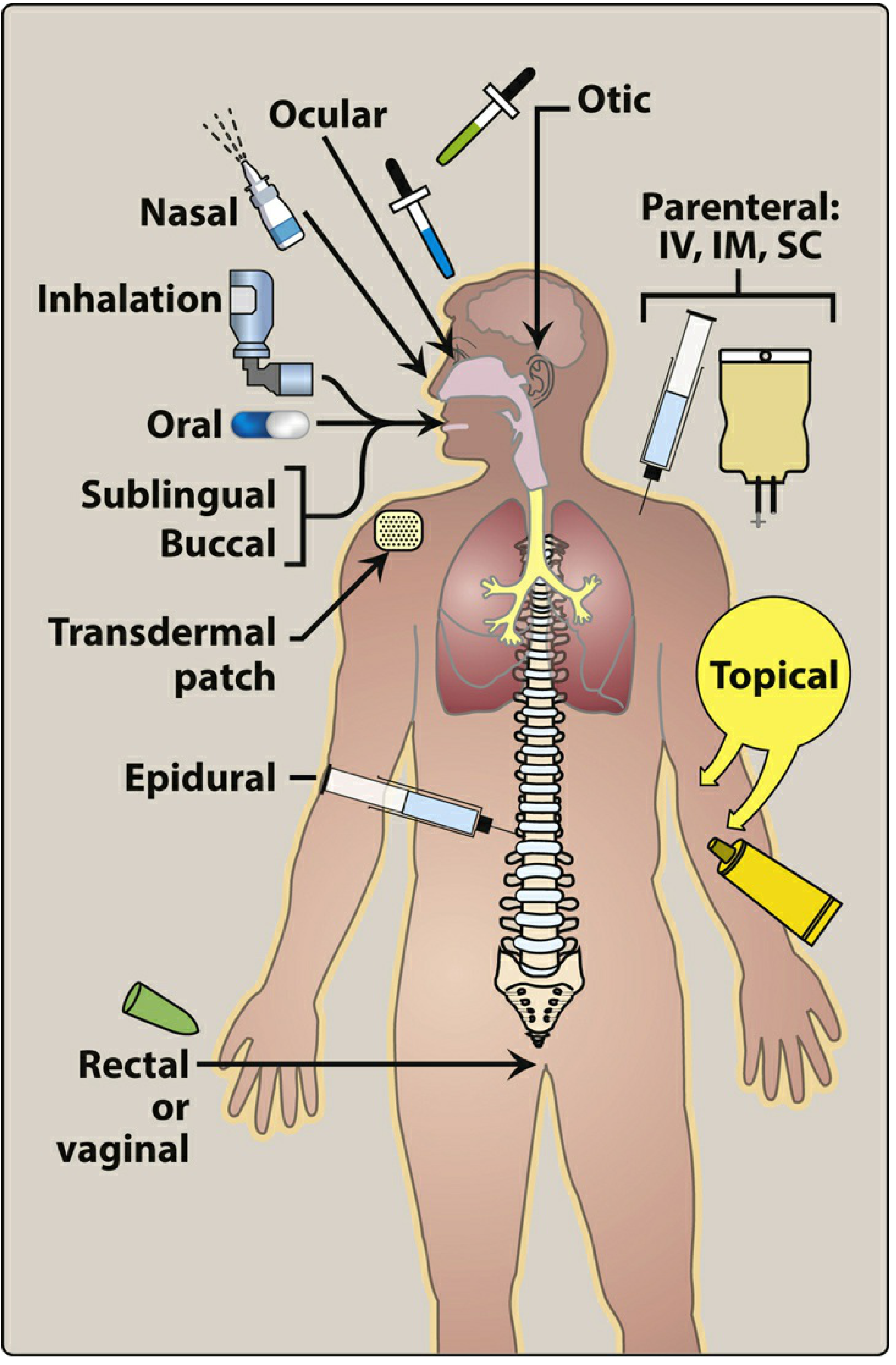
Lippincott Illustrated Reviews: Pharmacology
The route of administration is determined by drug properties (water/lipid solubility, ionization) and therapeutic objectives (speed of onset, duration, site specificity).
A. Enteral (via GI tract)
| Route | Description | Advantages | Disadvantages |
|---|---|---|---|
| Oral | Swallowed; absorbed from GI tract | Most common, convenient, economical | First-pass effect; slower onset; inactivated by stomach acid |
| Sublingual | Placed under tongue | Rapid absorption; bypasses first-pass metabolism | Limited drug capacity; must be lipid-soluble |
| Buccal | Between gum and cheek | Sustained release possible | Inconvenient |
Dental relevance: Sublingual and buccal routes are important in dentistry - e.g., nitroglycerine (angina emergency), midazolam (anxiolysis). Buccal mucosa also serves as a site for local drug delivery in periodontics.
B. Parenteral (bypasses GI tract)
| Route | Onset | Uses |
|---|---|---|
| Intravenous (IV) | Instantaneous | Emergencies, precise dosing |
| Intramuscular (IM) | 10-30 min | Depot preparations, vaccines |
| Subcutaneous (SC) | 15-30 min | Insulin, vaccines |
| Intra-articular | Local | Corticosteroid injections |
| Intraosseous | Rapid | Emergency when IV unavailable |
Dental relevance: Local anesthetic infiltration and nerve blocks in dentistry are a form of parenteral administration. Intraosseous anesthesia is a specific technique used in dental practice.
C. Topical/Local
- Applied directly to skin or mucous membranes
- Advantages: Localized action, minimal systemic side effects
- Dental relevance: Topical anesthetics (benzocaine gel), chlorhexidine mouthwash, antifungal oral gels
D. Inhalation
- Rapid absorption via large alveolar surface area
- Used for: General anesthetics (nitrous oxide in dentistry), bronchodilators
4. Pharmacokinetics - ADME
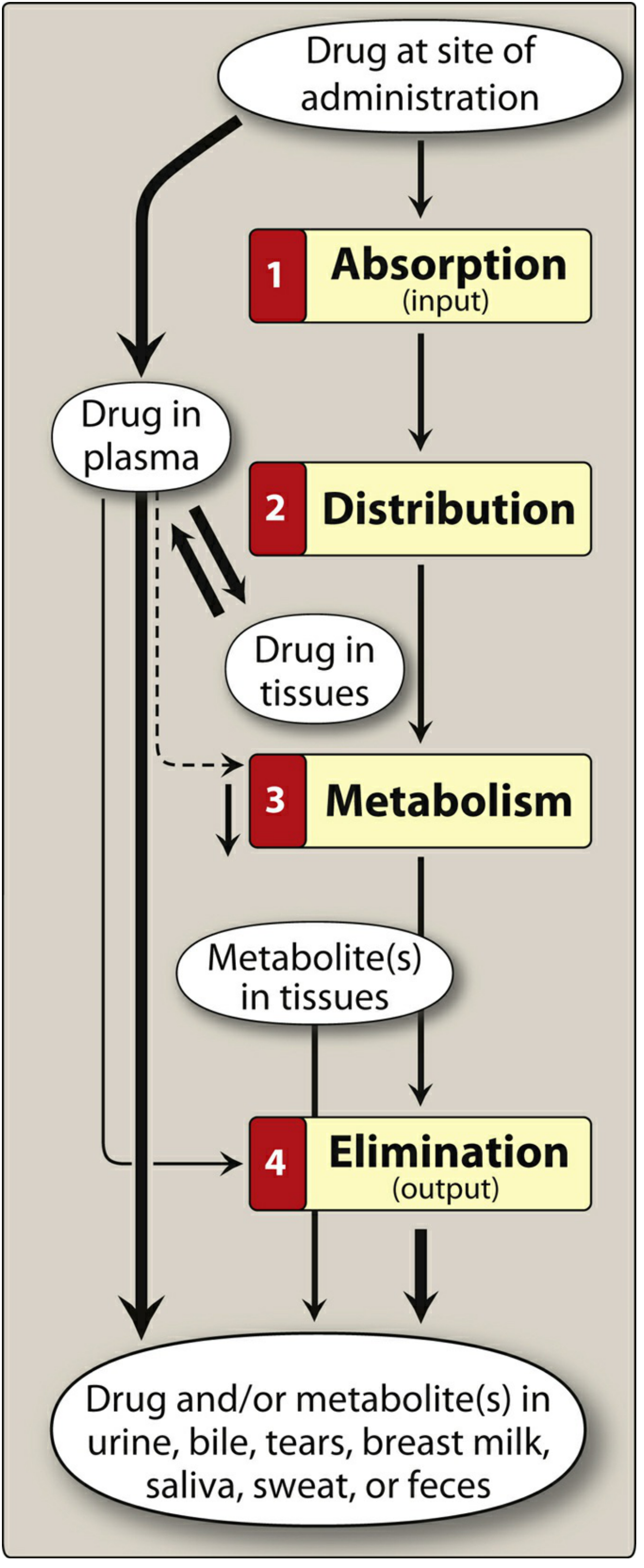
Lippincott Illustrated Reviews: Pharmacology
A. Absorption
Definition: Movement of a drug from its site of administration into the systemic circulation.
Mechanisms of Drug Transport Across Membranes
- Passive diffusion - most common mechanism; drug moves from high to low concentration; no carrier required; not saturable; lipid-soluble drugs cross most membranes easily
- Facilitated diffusion - carrier-mediated; no energy required; can be saturated; competitive inhibition possible
- Active transport - carrier-mediated; energy (ATP) dependent; moves drug against concentration gradient; saturable and selective
- Endocytosis - for very large molecules (e.g., Vitamin B12 absorbed via endocytosis in gut wall)
Effect of pH on Drug Absorption (Henderson-Hasselbalch)
- Most drugs are weak acids or weak bases
- Uncharged (un-ionized) form penetrates cell membranes readily
- For weak acids (e.g., aspirin, penicillin): un-ionized in acidic environment - better absorbed in stomach
- For weak bases (e.g., morphine, local anesthetics like lidocaine): un-ionized in alkaline environment - better absorbed in intestine
- pKa: lower pKa = more acidic drug; higher pKa = more basic drug
Clinical pearl for BDS: Local anesthetics (weak bases, pKa ~7.8-9.0) work poorly in infected tissues because the acidic environment of infection keeps them ionized, reducing penetration to nerve membranes. This is why dental blocks can fail in acute dental abscesses.
Bioavailability
- Definition: Fraction of administered drug that reaches systemic circulation unchanged
- IV route = 100% bioavailability
- Oral bioavailability is reduced by: first-pass metabolism, poor GI absorption
- First-pass (hepatic) effect: Drug absorbed from GI tract passes through portal vein to liver before reaching systemic circulation. Extensive hepatic metabolism here reduces bioavailability (e.g., lidocaine has very high first-pass effect, which is why it cannot be given orally as an antiarrhythmic - relevant in dentistry when considering systemic effects of injected lidocaine)
B. Distribution
Definition: Reversible movement of a drug from the systemic circulation into tissues and organs.
Factors Affecting Distribution
-
Plasma protein binding
- Albumin is the major drug-binding protein
- Only free (unbound) drug is pharmacologically active and can cross membranes
- Bound drug = inactive reservoir; dissociates as free drug is eliminated
- Drug interactions occur when two drugs compete for the same plasma protein binding sites
-
Tissue binding
- Some drugs accumulate in tissues (higher tissue than plasma concentration)
- Tissue reservoirs can prolong drug action
-
Lipophilicity
- Lipophilic drugs cross cell membranes freely; distribution governed mainly by blood flow
- Hydrophilic drugs pass through slit junctions only; confined to extracellular space
-
Blood-Brain Barrier (BBB)
- Tight junctions of brain capillaries limit entry of hydrophilic drugs
- Lipophilic drugs and those that are substrates for transport proteins cross more readily
Volume of Distribution (Vd)
Vd = Amount of drug in body / Plasma drug concentration at time zero (C₀)
| Vd (approx.) | Distribution pattern | Example |
|---|---|---|
| ~4 L | Plasma only (high MW or high protein binding) | Heparin |
| ~14 L | Extracellular fluid | Aminoglycosides |
| ~42 L (60% body weight) | Total body water | Ethanol |
| Very large (>100 L) | Extensive tissue binding | Chloroquine |
- A large Vd = extensive tissue distribution
- A small Vd = confined to plasma
C. Metabolism (Biotransformation)
Primary site: Liver (also intestinal wall, lung, plasma, kidney)
Purpose: Convert lipophilic drugs into more polar (water-soluble) metabolites for excretion
Phase I Reactions - "Functionalization"
- Add or expose a functional group (-OH, -NH₂, -SH, -COOH)
- Main system: Cytochrome P450 (CYP450) enzyme system in liver microsomes
- Products may be active, inactive, or toxic
Important CYP450 Isozymes for Dentistry:
| Isozyme | Substrates (dental relevance) | Inducers | Inhibitors |
|---|---|---|---|
| CYP2C9 | NSAIDs (ibuprofen), warfarin | Rifampin, phenobarbital | Fluconazole |
| CYP2D6 | Codeine (pro-drug - activated by 2D6) | None significant | Fluoxetine |
| CYP3A4 | Erythromycin, nifedipine, carbamazepine | Rifampin, phenytoin | Ketoconazole, erythromycin |
Dental relevance: Ketoconazole (antifungal used in oral candidiasis) inhibits CYP3A4, increasing concentrations of many co-administered drugs including some antihistamines and corticosteroids. Erythromycin also inhibits CYP3A4.
Phase II Reactions - "Conjugation"
- Attach (conjugate) polar molecule to drug or Phase I metabolite
- Common conjugates: Glucuronic acid (most common), sulfate, acetyl group, amino acid
- Result: Polar, water-soluble, typically inactive metabolite excreted in urine or bile
- Exception: Morphine-6-glucuronide is MORE potent than morphine
Enzyme Induction:
- Some drugs increase CYP enzyme synthesis (e.g., rifampin, phenobarbital, phenytoin, carbamazepine)
- Result: Faster metabolism of co-administered drugs, reducing their plasma levels
- Example: A patient on carbamazepine (for trigeminal neuralgia) will metabolize many drugs faster
Enzyme Inhibition:
- Some drugs decrease CYP activity
- Result: Increased plasma concentration of co-administered drugs - risk of toxicity
- Important inhibitors: Ketoconazole, clarithromycin, ritonavir (each inhibit multiple CYP isozymes)
D. Elimination (Excretion)
Primary route: Kidney (urine)
Other routes: Bile/feces, saliva, sweat, breast milk, tears, expired air
Renal Excretion
Three processes:
- Glomerular filtration - free (unbound) drug is filtered; protein-bound drug is not
- Active tubular secretion - active transport into tubular lumen; handles both free and protein-bound drug
- Passive tubular reabsorption - un-ionized/lipophilic drugs are reabsorbed back; ion trapping can promote excretion
Ion trapping: Alkalinizing urine (sodium bicarbonate) promotes excretion of weak acids (e.g., aspirin, phenobarbital) because they become more ionized in alkaline urine and cannot be reabsorbed.
This explains why sodium bicarbonate is given in aspirin overdose (as in the Katzung case study opening).
5. Key Pharmacokinetic Parameters
Half-Life (t½)
- Time required for plasma drug concentration to fall by 50%
- t½ = 0.693 × Vd / Clearance
- After 4-5 half-lives: drug is ~94-97% eliminated (clinically considered eliminated)
- Steady state is reached after approximately 4-5 half-lives of repeated dosing
- 90% of steady state is achieved in 3.3 half-lives
Clearance (CL)
- Volume of plasma cleared of drug per unit time
- CL = Vd × 0.693 / t½
Steady State
- Plasma concentration at which the rate of drug administration equals the rate of elimination
- Loading dose can be used to achieve steady state rapidly (important in emergencies)
6. Pharmacodynamics - Mechanisms of Drug Action
Drug Receptors
A receptor must:
- Selectively bind specific ligands (selectivity)
- Change function upon binding to alter biologic system activity
Types of Receptors
| Type | Location | Mechanism | Examples |
|---|---|---|---|
| Ligand-gated ion channels | Cell membrane | Direct ion channel opening | Nicotinic receptors, GABA-A |
| G-protein coupled receptors (GPCRs) | Cell membrane | Activate/inhibit intracellular enzymes via G-protein | β-adrenoceptors, muscarinic receptors, opioid receptors |
| Enzyme-linked receptors | Cell membrane | Receptor is or activates an enzyme (often tyrosine kinase) | Insulin receptor, growth factors |
| Intracellular (nuclear) receptors | Cytoplasm/nucleus | Gene transcription changes | Glucocorticoids, thyroid hormones |
Drug-Receptor Interactions
| Term | Definition |
|---|---|
| Agonist | Binds receptor and activates it (produces a response) |
| Full agonist | Can produce maximum possible response (100% efficacy) |
| Partial agonist | Binds and activates receptor but cannot produce maximum response, even at saturating concentrations; can act as antagonist in presence of full agonist |
| Antagonist | Binds receptor but does NOT activate it; blocks agonist access |
| Competitive antagonist | Competes with agonist at same receptor site; effect overcome by increasing agonist concentration |
| Non-competitive antagonist | Binds irreversibly or at allosteric site; maximum response cannot be overcome |
| Inverse agonist | Binds receptor and produces OPPOSITE effect to agonist (decreases constitutive activity) |
Dose-Response Relationships
- Efficacy (Emax): Maximum response a drug can produce
- Potency: Amount of drug needed to produce a given effect (usually ED50 - dose producing 50% of max effect)
- A drug can be potent but low efficacy, or vice versa
Therapeutic Index (TI)
TI = TD50 / ED50 (or LD50 / ED50)
- TD50 = dose causing toxicity in 50% of subjects
- ED50 = dose effective in 50% of subjects
- Narrow TI drugs (e.g., digoxin, lithium, warfarin, aminoglycosides, phenytoin) require careful dose titration and plasma monitoring
7. Adverse Drug Reactions (ADRs)
| Type | Description | Example |
|---|---|---|
| Type A (Augmented) | Predictable, dose-dependent extension of pharmacologic effect | NSAIDs causing gastric ulcer |
| Type B (Bizarre) | Unpredictable, not dose-dependent, often immunological | Penicillin anaphylaxis |
| Type C (Chronic) | Long-term effects | Corticosteroid osteoporosis |
| Type D (Delayed) | Appear after long latency | Drug-induced teratogenesis |
Drug Allergy:
- Immune-mediated reaction (Type B ADR)
- Types: IgE-mediated (anaphylaxis), cytotoxic (Type II), immune complex (Type III), T-cell mediated (Type IV)
- Dental relevance: Penicillin and local anesthetics (especially esters like procaine) can cause allergic reactions; always take drug allergy history before prescribing
8. Drug Interactions
Pharmacokinetic Interactions
- Absorption: Antacids chelate tetracyclines; reduce absorption
- Metabolism: Enzyme inducers (rifampin) reduce efficacy; inhibitors (ketoconazole) increase toxicity
- Protein binding displacement: One drug displaces another from albumin, increasing free drug levels (e.g., NSAIDs + warfarin)
- Excretion: Probenecid blocks tubular secretion of penicillin (used therapeutically to prolong penicillin effect)
Pharmacodynamic Interactions
- Synergism: Two drugs with similar effects - additive or supraadditive response
- Antagonism: One drug diminishes effect of another (e.g., naloxone reverses opioid effect)
9. Special Topics Relevant to Dentistry
Local Anesthetics (Dental Pharmacology Application)
- All commonly used dental local anesthetics are amide-linked (lidocaine, mepivacaine, articaine, bupivacaine, prilocaine)
- Mechanism: Block voltage-gated sodium channels in nerve membranes, preventing depolarization
- Work better in non-inflamed tissue (higher pH allows un-ionized form to penetrate)
- Metabolized in liver (amides) - use with caution in liver disease
- Vasoconstrictors (epinephrine, felypressin) added to:
- Reduce systemic absorption of local anesthetic
- Prolong duration of anesthesia
- Reduce bleeding in surgical field
Drugs with Dry Mouth (Xerostomia) as Side Effect
Significant in dentistry as xerostomia increases caries risk:
- Anticholinergics (atropine, antihistamines)
- Antihypertensives (diuretics, beta-blockers)
- Antidepressants (tricyclics, SSRIs)
- Antipsychotics
- Opioid analgesics
Drug-Induced Gingival Enlargement
Caused by:
- Phenytoin (anti-epileptic) - most classic
- Cyclosporin (immunosuppressant)
- Nifedipine and calcium channel blockers
Mechanism: Fibroblast proliferation with increased collagen production.
10. Prescription Writing
A valid prescription contains:
- Superscription: Date, patient's name and address, Rx symbol
- Inscription: Drug name, strength, and form
- Subscription: Quantity to dispense
- Signatura (Sig): Directions for patient
- Prescriber's signature and DEA number (for controlled substances)
Key abbreviations:
| Abbreviation | Meaning |
|---|---|
| OD | Once daily |
| BD/BID | Twice daily |
| TDS/TID | Three times daily |
| QID | Four times daily |
| AC | Before meals |
| PC | After meals |
| HS | At bedtime |
| PRN | As needed |
| Stat | Immediately |
| PO | By mouth (per os) |
11. Drug Nomenclature
| Name Type | Description | Example |
|---|---|---|
| Chemical name | IUPAC chemical structure | N-(4-hydroxyphenyl)acetamide |
| Generic (non-proprietary) name | Official approved name (INN) | Paracetamol / Acetaminophen |
| Brand (proprietary) name | Manufacturer's trade name | Tylenol, Calpol |
Summary: Key Concepts at a Glance
| Topic | Key Point |
|---|---|
| Pharmacokinetics | ADME - Absorption, Distribution, Metabolism, Elimination |
| Phase I metabolism | CYP450 enzymes - oxidation, reduction, hydrolysis |
| Phase II metabolism | Conjugation (glucuronidation most common) |
| pKa and absorption | Un-ionized drug crosses membranes; weak acids absorbed in acidic pH, weak bases in alkaline pH |
| Bioavailability | Fraction reaching systemic circulation; IV = 100%; oral reduced by first-pass effect |
| Half-life | Time for 50% reduction in plasma concentration; steady state after 4-5 t½ |
| Volume of distribution | Apparent fluid volume containing all drug at same plasma concentration |
| Receptor agonist | Binds + activates receptor |
| Receptor antagonist | Binds but does NOT activate; blocks agonist |
| Therapeutic index | TI = TD50/ED50; narrow TI drugs need monitoring |
| Local anesthetic failure | Acidic infected tissue keeps LA ionized, impairing nerve penetration |
| Drug-induced gingival enlargement | Phenytoin, cyclosporin, nifedipine |
Sources: Lippincott Illustrated Reviews: Pharmacology (9781975170561) | Katzung's Basic and Clinical Pharmacology 16e (9781260463309) | Goodman & Gilman's The Pharmacological Basis of Therapeutics (9781264258079)
This is a shared conversation. Sign in to Orris to start your own chat.