Section 7 in Harrison’s alterations in the renal and urinary tract function, chapter number 54, interstitial/bladder pain syndrome, chapter number 55, rcr unit abnormalities, chapter 56, fluid, electro disturbances, chapter 57, hypercalcaemia hypocalcaemia, chapter 58, and alkalosis, teach me everything to become the best doctor from Harrison
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Here is your complete Harrison's Section 7 master teaching guide - everything you need to become the best doctor:
Harrison's Section 7: Alterations in Renal & Urinary Tract Function
Complete Master Teaching Notes (Chapters 54-58)
CHAPTER 54 - Interstitial Cystitis / Bladder Pain Syndrome (IC/BPS)
Harrison's Principles of Internal Medicine 22E (2025), p. 377-380
What It Is
IC/BPS is a chronic pain disorder characterized by:
- Pelvic pain that worsens with bladder filling
- Associated urinary frequency and urgency
- Pain inside and/or outside the pelvis
- No identifiable infectious or structural cause
The diagnosis is frequently missed for years because physicians route patients by their most bothersome symptom - gynecologists (pelvic pain with menstrual flares), gastroenterologists (abdominal pain + bowel dysfunction), rheumatologists (generalized muscle/joint pain + fatigue), or internists (treated erroneously for recurrent UTIs).
Key Concept: Only 20-25% Have Bladder-Only Disease
This is the most important clinical pearl. Most IC/BPS patients have co-occurring pelvic conditions (COPCs) and systemic sensitization. You must treat the entire patient, not just the bladder. Four complicating factors:
- Spinal crosstalk - nerves from different pelvic organs converge
- Phenotype progression - disease evolves over time
- Central sensitization - amplified pain processing in CNS
- COPCs - co-occurring conditions (IBS, fibromyalgia, endometriosis, vulvodynia)
Primary Care Workup (Table 54-1)
| Step | What To Do |
|---|---|
| History + Physical | Pelvic exam including pelvic floor evaluation; categorize symptoms as bladder/pelvic vs. extending beyond pelvis |
| Urine Studies | Microscopic urinalysis + urine culture with sensitivity |
| Initial Treatment | Begin conservative measures if diagnosis is reasonably made |
| Referral Triggers | Unclear diagnosis; hematuria (microscopic or gross); refractory to treatment; severe or complex presentation |
Clinical Phenotypes - The "Clinical Picture" Approach
When patients present, classify their phenotype to guide therapy:
- Infection phenotype - recurrent UTIs triggering flares (treat each UTI; consider UTI vaccination)
- Pelvic floor phenotype - pelvic floor dysfunction on exam
- Psychosocial phenotype - depression, anxiety, PTSD, pain catastrophization = worse outcomes
- Systemic/COPC phenotype - fibromyalgia, IBS, widespread pain
Treatment Pyramid
1. Conservative Measures (First Line - All Patients)
- Patient education - acknowledge suffering, set realistic expectations (cure is not the goal), explain the chronic pain disorder model
- Dietary modifications - elimination diet for triggers: acidic/spicy foods, caffeine, alcohol, artificial sweeteners, gluten; highly individual
- Pelvic floor physiotherapy - randomized evidence supports this when pelvic floor dysfunction is present
- Stress management - sleep hygiene, exercise
2. Oral Medications
- Pentosan polysulfate (PPS) - only FDA-approved oral therapy; glycosaminoglycan (GAG) layer replenishment; note: retinal toxicity risk with prolonged use
- Amitriptyline - low-dose TCA for pain modulation
- Hydroxyzine - antihistamine (mast cell stabilization)
- Cimetidine - H2 blocker (some evidence)
- Quercetin - anti-inflammatory supplement
3. Intravesical Therapies (specialist)
- DMSO (dimethyl sulfoxide) - anti-inflammatory, analgesic
- Heparin/lidocaine/bicarbonate cocktails - GAG layer repair + local anesthesia
4. Procedural/Surgical (specialist)
- Cystoscopy with hydrodistension - diagnostic and modestly therapeutic
- Botulinum toxin A (BTX-A) injections
- Sacral neuromodulation (InterStim)
- Cyclosporine A - for refractory cases
- Surgery - cystectomy with urinary diversion only for extreme refractory cases
Psychological Interventions
- Cognitive behavioral therapy (CBT)
- Mindfulness-based stress reduction
- Pain psychologists - critical for psychosocial phenotype
CHAPTER 55 - Renal/Urine Unit Abnormalities (Hematuria, Proteinuria, Pyuria, Renal Tubular Disorders)
Harrison's Principles of Internal Medicine 22E (2025), block34
HEMATURIA
Definition
- Microscopic hematuria = ≥3-5 RBCs per high-power field (HPF) in spun first-morning urine
- Gross hematuria = visible blood in urine
Glomerular vs. Non-glomerular Hematuria
| Feature | Glomerular | Non-glomerular |
|---|---|---|
| RBC morphology | Dysmorphic RBCs | Normal/isomorphic |
| Casts | RBC casts | Absent |
| Protein | Usually elevated | May be absent |
| Causes | IgA nephropathy, GN, vasculitis, lupus | Stones, malignancy, BPH, cysts, papillary necrosis |
Clinical Rule: RBC casts or dysmorphic RBCs = glomerulonephritis until proven otherwise
Workup of Microscopic Hematuria
- Exclude anatomic lesions - especially urinary tract malignancy in older men
- Think also: BPH, interstitial nephritis, papillary necrosis, hypercalciuria, kidney stones, cystic disease, renal vascular injury
- If RBC casts present - nephrology referral, renal biopsy likely needed
PROTEINURIA
Classification by Amount
| Level | Amount | Significance |
|---|---|---|
| Normal | ~8-10 mg albumin/24h | Physiologic |
| Microalbuminuria | 30-300 mg/24h | Early nephropathy (e.g., early diabetic nephropathy) |
| Frank proteinuria | >300 mg/24h | Significant kidney disease |
| Nephrotic range | >3.5 g/24h | Glomerular disease |
- Spot urine albumin/creatinine ratio (UACR) >30 mg/g triggers further investigation
- Benign (functional/transient) proteinuria: <1 g/24h, non-sustained; causes include fever, exercise, obesity, sleep apnea, emotional stress, CHF
- Orthostatic proteinuria: only upright; benign prognosis
- Selective proteinuria (albumin-only) = Minimal Change Disease in children
- Non-selective proteinuria = most adult glomerular diseases
PYURIA
- Leukocytes in urine
- Inflammatory glomerular disease (post-streptococcal GN, MPGN) can cause sterile pyuria
- Must distinguish from bacterial infection - culture is key
CLINICAL SYNDROMES FROM GLOMERULAR INJURY
| Syndrome | Features |
|---|---|
| Acute Nephritic | 1-2 g/24h proteinuria + hematuria + RBC casts + hypertension + elevated creatinine + fluid retention |
| Rapidly Progressive GN (RPGN) | Creatinine rises over days; crescentic GN on biopsy; causes: Goodpasture's, ANCA vasculitis, lupus |
| Nephrotic Syndrome | >3.5 g/24h proteinuria + hypoalbuminemia + edema + hyperlipidemia + lipiduria |
| Chronic GN | Slow progression to ESRD over years |
RENAL TUBULAR TRANSPORT ABNORMALITIES (Ch 55)
The renal tubule handles sodium, potassium, bicarbonate, glucose, amino acids, phosphate, urate, and water through highly specialized transporters. Defects in these transporters produce specific clinical syndromes:
Key Tubular Syndromes
Proximal Tubule (PCT)
- Fanconi syndrome - generalized PCT dysfunction: glycosuria (normal blood glucose), aminoaciduria, phosphaturia, uricosuria, bicarbonaturia → hypokalemia, metabolic acidosis, hypophosphatemia
- Causes: cystinosis, Wilson's disease, galactosemia, tenofovir toxicity, heavy metals (lead, cadmium), multiple myeloma
Distal Tubule / Collecting Duct
- Renal Tubular Acidosis (RTA) - see Ch 58 content below
- Nephrogenic Diabetes Insipidus (NDI) - ADH resistance: massive polyuria/polydipsia with dilute urine, serum sodium often normal/elevated; causes: lithium, hypercalcemia, hypokalemia, congenital (aquaporin-2 mutations)
- Bartter Syndrome - loss-of-function mutations in Na-K-2Cl cotransporter (NKCC2) in thick ascending limb of Henle → hypokalemia, metabolic alkalosis, hyperreninism, hyperaldosteronism, normal blood pressure
- Gitelman Syndrome - loss-of-function mutation of Na-Cl cotransporter (NCC) in DCT → hypokalemia, metabolic alkalosis, hypomagnesemia, hypocalciuria; milder than Bartter's
- Liddle's Syndrome - gain-of-function mutation of ENaC (collecting duct Na channel) → hypertension, hypokalemia, metabolic alkalosis, low renin, low aldosterone; treated with amiloride or triamterene (not spironolactone)
CHAPTER 56 - Fluid and Electrolyte Disturbances in CKD
Harrison's Principles of Internal Medicine 22E (2025), p. 220+
Sodium and Water Homeostasis in CKD
Key mechanism: With declining GFR, many CKD patients retain sodium because excretion can no longer match intake. This causes:
- Extracellular fluid volume (ECFV) expansion
- Hypertension → accelerates nephron hyperfiltration and injury
- Isonatric expansion (plasma sodium often NORMAL) as long as water doesn't exceed clearance capacity
- Hyponatremia is uncommon in CKD but responds to water restriction
Management of Sodium/Water Excess
- Salt restriction (dietary counseling)
- Thiazides (chlorthalidone) - effective even in stage 4 CKD for BP control
- Loop diuretics (furosemide, bumetanide, torsemide) - needed for frank sodium accumulation; resistance common in CKD → use higher doses
- Combination therapy: loop diuretic + metolazone for diuretic resistance
- Intractable edema + hypertension in advanced CKD = indication to initiate dialysis
The Volume Depletion Trap
CKD kidneys cannot conserve sodium well either. With extrarenal losses (GI losses, over-diuresis):
- Leads to prerenal acute-on-chronic kidney failure
- Action: hold diuretics ± cautious NS repletion
- "Sick day rules" - tell patients to hold diuretics/antihypertensives if vomiting/diarrhea
Potassium Homeostasis in CKD
Paradox: GFR decline is NOT matched by equal decline in K+ excretion because:
- Aldosterone-dependent secretion in distal nephron compensates
- Augmented GI K+ excretion compensates
Causes of Hyperkalemia in CKD
- High dietary K+ intake
- Hemolysis, stored blood transfusion
- Metabolic acidosis (K+ shifts out of cells)
- Medications (most important):
- RAS inhibitors (ACEi, ARB)
- Spironolactone, eplerenone, amiloride, triamterene
- Nonsteroidal mineralocorticoid receptor antagonists (finerenone)
- NSAIDs
- TMP-SMX (blocks ENaC like amiloride)
Clinical balance: RAS inhibitors' benefits (renal protection, cardioprotection) often outweigh hyperkalemia risk - use with close monitoring; co-administer potassium binders (patiromer, sodium zirconium cyclosilicate) to allow RAS inhibitor use
SGLT2 inhibitors (gliflozins): even in advanced CKD, appear to have counterbalancing effects on K+ that result in net preservation of K+ homeostasis - favorable profile
Special CKD Causes of Severe Hyperkalemia
Conditions with disproportionate loss of distal K+ secretion mechanisms:
- Hyporeninemic hypoaldosteronism (type 4 RTA) - especially in diabetic nephropathy
- Obstructive uropathy
- Sickle cell nephropathy
Acid-Base in CKD
- Stage 3B CKD: hyperchloremic (normal AG) metabolic acidosis due to impaired NH4+ excretion
- Stage 4-5 CKD: transitions to high-AG metabolic acidosis (uremic anion accumulation)
- Treatment: oral alkali replacement (NaHCO3 tablets 650 mg = 7.8 mEq; or Shohl's solution [sodium citrate]) to maintain HCO3- >22-24 mmol/L
- Benefits: slows CKD progression, reduces muscle catabolism, reduces bone disease
- Fruits and vegetables (dietary alkali/citrate) can raise HCO3- and slow CKD progression safely
CHAPTER 57 - Hypercalcemia and Hypocalcemia
Harrison's Principles of Internal Medicine 22E (2025), p. 262+
Calcium Physiology - The Regulation System
Normal serum calcium: 8.5-10.5 mg/dL (2.1-2.6 mM)
The feedback system involves 4 organs and 2 hormones:
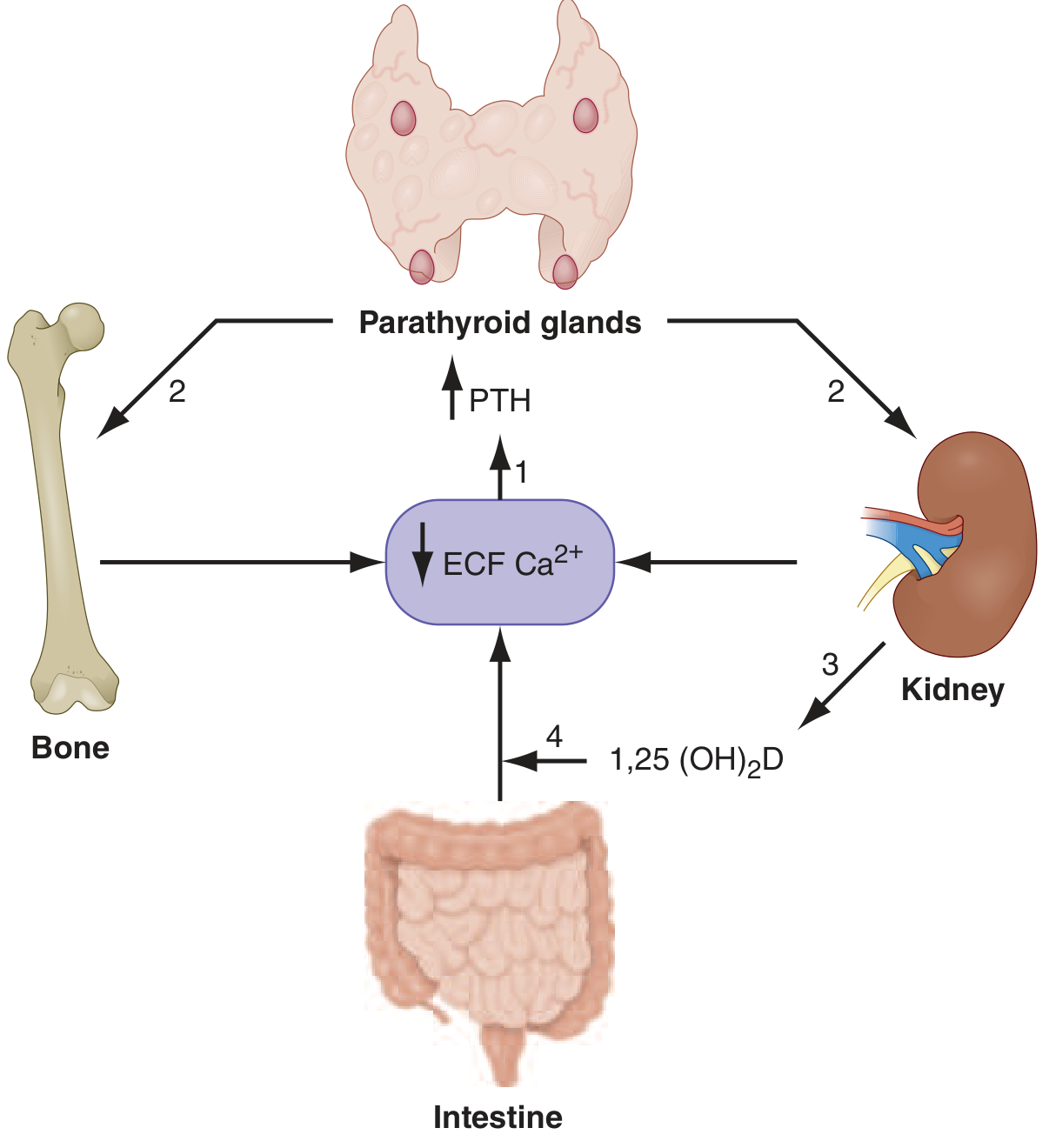
PTH response to low calcium (via CaSR on parathyroid chief cells):
- Increases osteoclast-mediated bone resorption (via RANKL on osteocytes) → releases Ca2+ from bone
- Increases distal tubule Ca2+ reabsorption in kidney
- Stimulates proximal tubule 1,25(OH)2D (calcitriol) production
- 1,25(OH)2D increases intestinal Ca2+ absorption
Calcium fractions in plasma:
- ~50% ionized (physiologically active)
- ~40% albumin-bound
- ~10% complexed to anions (citrate, phosphate)
Corrected calcium formula: Add 0.8 mg/dL for every 1 g/dL albumin is below 4.0 g/dL
(or: corrected Ca = measured Ca + 0.8 × [4.0 - albumin])
HYPERCALCEMIA
Causes (Table 57-1) - Organized by Mechanism
1. Excess PTH (PTH elevated or inappropriately normal)
- Primary hyperparathyroidism (pHPT) - #1 most common cause overall; parathyroid adenoma (~85%), hyperplasia (~15%), carcinoma (<1%); often asymptomatic, found incidentally
- Sporadic or familial (MEN1, MEN2A)
- ~30% of sporadic pHPT have genetic cause (cyclin D1 or menin)
- ~50% have reduced CaSR expression due to epigenetic silencing
- Tertiary hyperparathyroidism - long-term PTH stimulation in renal failure → autonomous PTH secretion persists even after transplant
- Familial Hypocalciuric Hypercalcemia (FHH) - inactivating CaSR mutations → impaired Ca2+ sensing in parathyroid AND kidney → mild hypercalcemia + normal or low urine calcium excretion + elevated PTH; benign, no treatment needed; distinguish from pHPT (24h urine Ca/creatinine clearance ratio <0.01 in FHH)
- Lithium therapy - impairs CaSR signaling
- Ectopic PTH secretion - very rare
2. Malignancy-associated (PTH suppressed)
- PTHrP (PTH-related peptide) - most common mechanism in solid tumors (lung, breast, head/neck, renal, bladder); PTHrP shares N-terminal homology with PTH and binds same receptor → "humoral hypercalcemia of malignancy"
- Lytic bone metastases - direct osteolytic destruction (breast cancer, multiple myeloma)
- Ectopic 1,25(OH)2D production - lymphomas, some solid tumors; macrophages in tumor express CYP27B1 constitutively
3. Granulomatous diseases (PTH suppressed, 1,25(OH)2D elevated)
- Sarcoidosis (#1), tuberculosis, leprosy, fungal infections (histoplasmosis, coccidioidomycosis), foreign body reactions, berylliosis
- Macrophage CYP27B1 converts 25(OH)D → 1,25(OH)2D without normal feedback regulation
- Treatment: glucocorticoids suppress macrophage 1α-hydroxylase
4. Vitamin D intoxication
- Excessive 25(OH)D ingestion overwhelms vitamin D binding protein (DBP), freeing 25(OH)D to directly activate intestinal VDR → hypercalcemia; can persist weeks (longer half-life of 25[OH]D vs. 1,25[OH]2D)
- CYP24A1 deficiency - genetic loss of 24-hydroxylase (inactivates 1,25[OH]2D) → 1,25(OH)2D accumulation
5. Bone-Related Causes
- Hyperthyroidism (T3/T4 increase osteoclast activity directly)
- Paget's disease with immobilization
- Immobilization alone (especially in Paget's or adolescents)
- Osteolytic metastases
6. Excess Calcium Intake
- Milk-alkali syndrome - calcium carbonate antacid overuse → hypercalcemia + metabolic alkalosis + renal failure (now more common with calcium supplements)
7. Medications
- Thiazides (increase renal Ca reabsorption)
- Lithium
- Vitamin A (excess)
- Teriparatide (PTH analog)
- Denosumab discontinuation → rebound resorption
Clinical Features ("Bones, Stones, Groans, Psychic Moans")
| System | Manifestations |
|---|---|
| Neuro/Psych | Weakness, fatigue, depression, cognitive impairment, coma (severe) |
| GI ("Groans") | Nausea, vomiting, constipation, peptic ulcer (PTH stimulates gastrin), pancreatitis |
| Renal ("Stones") | Nephrolithiasis (Ca-oxalate, Ca-phosphate), nephrocalcinosis, polyuria/polydipsia (NDI), renal tubular acidosis |
| Bone ("Bones") | Bone pain, osteitis fibrosa cystica (in severe pHPT: brown tumors, subperiosteal bone resorption), osteoporosis, pathologic fractures |
| CV | Shortened QT interval, bradycardia, hypertension |
Diagnosis
- PTH level is the first branch point:
- PTH elevated/inappropriately normal → primary or tertiary HPT, FHH, lithium
- PTH suppressed → malignancy, vitamin D toxicity, granulomatous disease, excess calcium intake
- PTHrP level - if PTH suppressed and malignancy suspected
- 1,25(OH)2D level - elevated in granulomatous disease and lymphoma
- 25(OH)D level - elevated in vitamin D intoxication
- 24h urine calcium - very low in FHH vs. high in pHPT
Treatment of Hypercalcemia
Acute hypercalcemic crisis (Ca >14 mg/dL or symptomatic):
- IV hydration with normal saline (2-4 L/day) - volume expansion increases GFR → calciuresis (MOST IMPORTANT first step)
- Loop diuretics (furosemide) - only AFTER volume repletion; promotes urinary Ca2+ excretion; do NOT use thiazides (they worsen hypercalcemia)
- Bisphosphonates (zoledronic acid 4 mg IV or pamidronate 60-90 mg IV) - inhibit osteoclast bone resorption; onset 24-72h, peak effect 4-7 days; mainstay for malignancy-associated hypercalcemia
- Calcitonin (4-8 IU/kg SC q6-12h) - rapid onset (hours), modest effect; tachyphylaxis within 48h; useful bridge while waiting for bisphosphonates to act
- Denosumab (anti-RANKL) - for bisphosphonate-refractory or bisphosphonate-contraindicated (renal failure) malignancy hypercalcemia
- Glucocorticoids (prednisone 40-60 mg/day or hydrocortisone 200-300 mg/day) - effective for granulomatous disease, lymphoma, vitamin D intoxication
- Hemodialysis - for life-threatening, refractory hypercalcemia with renal failure
Surgery (cinacalcet for medical Rx):
- Parathyroidectomy for symptomatic pHPT or asymptomatic with criteria (Ca >1 mg/dL above upper normal, T-score <-2.5, age <50, GFR <60, 24h urine Ca >400 mg)
- Cinacalcet (calcimimetic - activates CaSR) - for patients not surgical candidates; lowers PTH and Ca without improving bone density; also for tertiary/secondary HPT
HYPOCALCEMIA
Causes (Table 57-2)
Reduced PTH (Hypoparathyroidism)
- Post-surgical (thyroidectomy, parathyroidectomy) - #1 acquired cause
- Autoimmune (autoimmune polyglandular syndrome type 1 - APS-1; associated with AIRE mutations)
- Activating CaSR mutations (autosomal dominant hypocalcemia - ADH; gain-of-function CaSR → inappropriately suppressed PTH)
- Hypomagnesemia - impairs both PTH secretion AND PTH action at target organs; must correct Mg first before Ca rises
- Infiltration (metastases, hemochromatosis, Wilson's, sarcoidosis)
- Hungry bone syndrome after parathyroidectomy (rapid bone uptake)
- Radiation
- Severe illness (sepsis, burns, pancreatitis - multifactorial)
PTH-resistant / Vitamin D Deficiency
- Vitamin D deficiency - dietary, malabsorption, lack of sun exposure, CKD (impaired 1α-hydroxylation), liver disease (impaired 25-hydroxylation), anti-epileptic drugs (phenytoin, phenobarbital - accelerate vitamin D catabolism)
- Pseudohypoparathyroidism (PHP) - Albright hereditary osteodystrophy; PTH is elevated but target organs (kidney, bone) are resistant (Gs-alpha mutation); PHP type 1a: Gs-alpha defect; short stature, brachydactyly, round face, ectopic calcifications
- Hypomagnesemia (PTH resistance)
Calcium Redistribution
- Acute pancreatitis (Ca2+ saponification)
- Rhabdomyolysis + renal failure (Ca2+ deposits in damaged muscle)
- Osteoblastic metastases (prostate cancer - rapid bone formation consumes Ca2+)
- Respiratory or metabolic alkalosis → increased albumin-Ca2+ binding → decreased ionized Ca2+
- Phosphate-containing enemas in renal disease → hyperphosphatemia → precipitation of Ca2+
Pseudohypocalcemia
- Low albumin (hypoalbuminemia from malnutrition, nephrotic syndrome, cirrhosis) - ionized Ca2+ is NORMAL; use corrected calcium formula
- Gadosetamide MRI contrast - interferes with colorimetric Ca assay
Clinical Features ("Chvostek, Trousseau, Tetany")
| System | Feature |
|---|---|
| Neuromuscular | Tetany, muscle cramps, paresthesias (perioral, fingertips), carpopedal spasm |
| Chvostek's sign | Tapping facial nerve → facial twitch (low specificity) |
| Trousseau's sign | Inflate BP cuff >systolic for 3 min → carpal spasm (more specific) |
| Severe | Laryngospasm, seizures, altered mental status, papilledema |
| Cardiac | Prolonged QT interval → risk of Torsades de Pointes |
| Chronic | Cataracts, dry skin/hair/nails, dental enamel defects, calcification of basal ganglia, calcification of sclera |
Treatment
Acute/Symptomatic Hypocalcemia (Tetany, Seizures, QT prolongation):
- IV Calcium gluconate (preferred over Ca chloride except in cardiac arrest due to less tissue necrosis if extravasation): 1-2 g IV over 10-20 min, then continuous infusion
- IV Calcium chloride in cardiac arrest (more bioavailable per mL)
- Correct magnesium deficiency first if Mg2+ is low (otherwise Ca2+ will not respond)
- Monitor cardiac rhythm
Chronic Hypocalcemia (Hypoparathyroidism):
- Calcium carbonate or citrate 1.5-3 g elemental Ca/day (citrate better absorbed with low gastric acid/PPIs)
- Calcitriol [1,25(OH)2D] 0.25-2 μg/day - since PTH is absent, 1α-hydroxylation is impaired → must give active form
- PTH replacement (Natpara/recombinant PTH 1-84) - for patients difficult to control
- Target: low-normal serum Ca, urine Ca <300 mg/day (to avoid nephrocalcinosis/nephrolithiasis)
- Thiazides paradoxically reduce urinary Ca2+ losses in hypoparathyroidism
CHAPTER 58 - Acidosis and Alkalosis
Harrison's Principles of Internal Medicine 22E (2025), p. 410-418
NORMAL ACID-BASE HOMEOSTASIS
Normal range: Arterial pH 7.35-7.45
Henderson-Hasselbalch Equation:
pH = 6.1 + log₁₀ (HCO3⁻) / (0.03 × PaCO2)
Two regulatory systems:
- Respiratory (CNS + lungs) - controls PaCO2 (normal ~40 mmHg); responds in minutes
- Renal (kidneys) - controls HCO3⁻ (normal 22-26 mmol/L); responds in hours-days
SYSTEMATIC APPROACH TO ACID-BASE DISORDERS
Step 1: Is it acidemia (pH <7.35) or alkalemia (pH >7.45)?
Step 2: What is the primary disorder?
| Disorder | pH | Primary Change | Compensation |
|---|---|---|---|
| Metabolic acidosis | ↓ | ↓HCO3⁻ | ↓PaCO2 (hyperventilate) |
| Metabolic alkalosis | ↑ | ↑HCO3⁻ | ↑PaCO2 (hypoventilate) |
| Respiratory acidosis | ↓ | ↑PaCO2 | ↑HCO3⁻ (renal) |
| Respiratory alkalosis | ↑ | ↓PaCO2 | ↓HCO3⁻ (renal) |
Step 3: Is the compensation appropriate? (Detect Mixed Disorders)
Winter's Equation (expected respiratory compensation for metabolic acidosis):
Expected PaCO2 = 1.5 × [HCO3⁻] + 8 ± 2
If measured PaCO2 is LOWER than expected → concurrent respiratory alkalosis
If measured PaCO2 is HIGHER than expected → concurrent respiratory acidosis
Metabolic compensation for respiratory disorders (Table 58-1):
- Acute respiratory acidosis: HCO3⁻ rises 1 mEq/L per 10 mmHg ↑PaCO2
- Chronic respiratory acidosis: HCO3⁻ rises 3.5 mEq/L per 10 mmHg ↑PaCO2
- Acute respiratory alkalosis: HCO3⁻ falls 2 mEq/L per 10 mmHg ↓PaCO2
- Chronic respiratory alkalosis: HCO3⁻ falls 5 mEq/L per 10 mmHg ↓PaCO2 (may fully normalize pH - exception to the rule)
- Metabolic alkalosis: PaCO2 rises 0.7 mmHg per 1 mEq/L ↑HCO3⁻ (limit ~55 mmHg)
Step 4: Calculate the Anion Gap
AG = Na⁺ - (Cl⁻ + HCO3⁻)
- Normal AG: 6-12 mmol/L (average ~10 mmol/L)
- ALWAYS correct for albumin: For each 1 g/dL albumin below 4.5 g/dL, add 2.5 mmol/L to reported AG
- Example: albumin 2.5 g/dL (2 g/dL below normal) + uncorrected AG 15 → corrected AG = 15 + (2 × 2.5) = 20 mmol/L
Unmeasured anions that contribute to normal AG: albumin, phosphate, sulfate, organic anions
High AG causes: MUDPILES + "GOLDMARK" mnemonic
| Mnemonic | Causes |
|---|---|
| Methanol | Formic acid accumulation |
| Uremia | Organic uremic anions |
| Diabetic ketoacidosis | Acetoacetate + beta-hydroxybutyrate |
| Propylene glycol | Drug vehicle (lorazepam, diazepam infusions) |
| Isoniazid / Iron | |
| Lactic acidosis | Type A (tissue hypoxia) or Type B (metformin, liver failure, malignancy) |
| Ethylene glycol | Oxalic acid accumulation |
| Salicylates | |
| Also: 5-Oxoproline | Acetaminophen overuse in malnourished/critically ill |
| Also: D-lactic acidosis | Short bowel syndrome; gut bacteria produce D-lactate |
Low AG causes:
- Increased unmeasured cations (hyperkalemia, hypercalcemia, hypermagnesemia)
- Abnormal cations: lithium intoxication, cationic immunoglobulins (myeloma)
- Reduced albumin (nephrotic syndrome, liver disease, malabsorption)
- Hyperviscosity, severe hyperlipidemia (underestimation of Na and Cl)
Step 5: In High-AG Metabolic Acidosis - Check the Delta-Delta (Δ/Δ)
Delta Ratio = ΔAG / ΔHCO3⁻ = (AG - 10) / (24 - HCO3⁻)
| Delta Ratio | Interpretation |
|---|---|
| <0.4 | Pure non-AG metabolic acidosis (hyperchloremic) |
| 0.4-1.0 | Mixed high-AG + non-AG metabolic acidosis |
| 1.0-2.0 | Pure high-AG metabolic acidosis |
| >2.0 | High-AG metabolic acidosis + coexisting metabolic alkalosis (or pre-existing elevated HCO3⁻) |
METABOLIC ACIDOSIS
High-AG Metabolic Acidosis
Lactic Acidosis:
- Type A: Tissue hypoxia (septic shock, cardiogenic shock, mesenteric ischemia, severe anemia, CO poisoning)
- Type B: No hypoxia (metformin toxicity, liver failure, malignancy, thiamine deficiency, propylene glycol, D-lactic acidosis)
- Normal lactate <2 mmol/L; >4 mmol/L = significant lactic acidosis
- Treatment: address underlying cause; bicarbonate controversial in lactic acidosis
Diabetic Ketoacidosis (DKA):
- Insulin deficiency + glucagon excess → unrestrained lipolysis → acetoacetate + beta-hydroxybutyrate
- Initially HAGMA; as treated, becomes mixed (non-AG hyperchloremic acidosis develops with saline infusion)
- Beta-hydroxybutyrate NOT detected by nitroprusside test (detects acetoacetate only) → can underestimate ketosis initially
Ethylene Glycol:
- Metabolized to oxalic acid → calcium oxalate crystals in urine (envelope-shaped)
- High osmolar gap early; high AG later (as metabolism occurs)
- Renal failure, cerebral edema, hypocalcemia
- Treatment: fomepizole (alcohol dehydrogenase inhibitor); dialysis
Methanol:
- Metabolized to formic acid → optic nerve toxicity (blindness)
- High osmolar gap early; high AG later
- Treatment: fomepizole; dialysis; folate (augments formate metabolism)
Salicylate Toxicity (Unique - Mixed Disorder):
- Direct stimulation of CNS chemoreceptors → respiratory alkalosis (early)
- Uncoupling of oxidative phosphorylation → metabolic acidosis (late)
- Classic presentation: high AG metabolic acidosis + respiratory alkalosis
- High osmolar gap typically absent
- Treatment: urine alkalinization (Na bicarbonate infusion) to trap ionized salicylate in urine; dialysis for severe cases
Isopropyl Alcohol:
- Metabolized to acetone (not an acid) → osmolar gap ELEVATED, but AG NORMAL (distinguishes from methanol/EG)
- Treatment: supportive; dialysis for severe coma, hemodynamic instability, or levels >400 mg/dL
Acetaminophen (Pyroglutamic acid / 5-oxoprolinemia):
- Acetaminophen + glutathione depletion → 5-oxoproline (pyroglutamic acid) accumulation
- Suspect in unexplained HAGMA WITHOUT elevated osmolar gap in malnourished/critically ill patients on acetaminophen
- Treatment: stop acetaminophen; IV NaHCO3; consider N-acetylcysteine
Non-Anion Gap (Hyperchloremic) Metabolic Acidosis
Mechanism: HCO3⁻ loss with reciprocal Cl⁻ gain; AG remains normal
Causes:
| Source | Causes |
|---|---|
| GI HCO3⁻ loss | Diarrhea (#1 most common cause worldwide), small bowel fistulas, ileal conduit, ileostomy, ureteral diversion, cholestyramine, pancreatic fistula |
| Renal HCO3⁻ loss / Failure to generate HCO3⁻ | Renal Tubular Acidosis (Types 1, 2, 4), early CKD (stage 3B) |
| Other | Saline infusion (dilutional), hyperalimentation, acetazolamide (carbonic anhydrase inhibition) |
Renal Tubular Acidosis (RTA) - Detailed
| Type | Defect | Urine pH | Urine AG | Serum K+ | Key Associations | Treatment |
|---|---|---|---|---|---|---|
| Type 1 (Distal) | H+ secretion defect in collecting duct; cannot acidify urine | >5.5 (urine stays alkaline) | Positive (+) | Low (hypokalemia) | Sjögren's syndrome, RA, lupus, hypercalciuria, nephrolithiasis (Ca-phosphate stones), amphotericin B, voltage-dependent (sickle cell, obstructive uropathy) | NaHCO3 or Na citrate |
| Type 2 (Proximal) | HCO3⁻ reabsorption defect in PCT; urinary bicarbonate wasting until new lower threshold | <5.5 (if acidotic, kidney eventually acidifies urine) | Negative (-) | Low (hypokalemia) | Fanconi syndrome, myeloma, carbonic anhydrase inhibitors, heavy metals, tenofovir, Wilson's | K+ citrate; higher alkali doses needed (HCO3⁻ leaks in urine); treat underlying cause |
| Type 3 | Combined type 1+2 features | Mixed | - | Low | Rare; carbonic anhydrase II deficiency | |
| Type 4 (Hyporeninemic hypoaldosteronism) | Aldosterone deficiency/resistance → impaired H+ and K+ secretion in collecting duct | <5.5 | Positive (+) | High (hyperkalemia) | Diabetic nephropathy (#1), obstructive uropathy, sickle cell, HIV nephropathy, NSAIDs, calcineurin inhibitors, heparin | Treat underlying cause; fludrocortisone; low-K diet; loop diuretics |
Urine Anion Gap (UAG) in non-AG metabolic acidosis:
UAG = Urine (Na⁺ + K⁺) - Urine Cl⁻
- Negative UAG = NH4⁺ high in urine → appropriate renal acid excretion → GI cause (diarrhea)
- Positive UAG = NH4⁺ low in urine → renal acid excretion failure → RTA
Urine osmolar gap can also quantify NH4⁺ excretion:
Urine osmolar gap = Measured urine osmolality - 2(Na+K) + glucose/18 + BUN/2.8
METABOLIC ALKALOSIS
Causes (Table 58-6)
I. ECFV depletion, normotension, K+ deficiency, low/normal renin-aldosterone:
- Gastrointestinal: Vomiting/NG suction (#1 cause) → HCl loss; pyloric stenosis in infants; congenital chloridorrhea
- Renal: Diuretics (thiazides, loop diuretics); post-hypercapnic state; recovery from lactic/ketoacidosis; non-reabsorbable anions (IV penicillin); Mg2+ deficiency; K+ depletion; Bartter's syndrome (TALH transporter mutations); Gitelman's syndrome (DCT Na-Cl cotransporter mutation)
II. ECFV expansion, hypertension, K+ deficiency, mineralocorticoid excess:
- High renin: renal artery stenosis, renin-secreting tumor, malignant hypertension, estrogen therapy
- Low renin/low aldosterone: Liddle's syndrome (ENaC gain-of-function)
- Low renin/high aldosterone: Primary aldosteronism (Conn's syndrome - adenoma or hyperplasia), 11β-hydroxylase/17α-hydroxylase deficiency, Cushing's syndrome
- Low renin/low aldosterone: Licorice (glycyrrhizin inhibits 11β-HSD2 → cortisol activates mineralocorticoid receptor), carbenoxolone
Pathophysiology of Vomiting-Induced Metabolic Alkalosis
- Generation phase: HCl loss from gastric juice → plasma HCO3⁻ rises; Na loss → ECFV depletion → secondary hyperaldosteronism; K+ loss → hypokalemia
- Maintenance phase: Volume depletion stimulates proximal HCO3⁻ reabsorption; hypokalemia drives K+/H+ exchange in collecting duct → paradoxical aciduria (kidney excretes acid despite alkalosis); hyperaldosteronism stimulates H+ secretion
Result: Hypochloremic, hypokalemic metabolic alkalosis with a urine pH that can be acidic ("paradoxical aciduria")
Urine Cl⁻ in Metabolic Alkalosis - Key Diagnostic Tool
| Urine Cl⁻ | Interpretation | Treatment |
|---|---|---|
| <25 mEq/L ("Saline-Responsive") | ECFV-depleted; chloride-deficient; kidneys avidly retaining Cl⁻ | IV NaCl + KCl correction; address underlying cause |
| >40 mEq/L ("Saline-Resistant") | Mineralocorticoid excess OR ongoing diuretic use; kidneys excreting Cl⁻ | Treat mineralocorticoid excess; spironolactone; amiloride |
Note: Urine pH is unreliable in metabolic alkalosis (can be acidic from paradoxical aciduria); use urine Cl⁻ instead
Treatment of Metabolic Alkalosis
- Saline-responsive (most common): IV NS + KCl to correct ECFV and chloride depletion; if due to diuretic use, discontinue diuretic
- Mineralocorticoid-mediated: Spironolactone (primary aldosteronism); amiloride or triamterene (Liddle's - ENaC blockers, NOT spironolactone); surgical (adenoma resection for Conn's); adrenalectomy or ketoconazole (Cushing's)
- Bartter's/Gitelman's: K+ supplements, K+-sparing diuretics, NSAIDs (indomethacin for Bartter's), Mg supplements for Gitelman's
- Vomiting-induced: Treat underlying cause; correct ECFV + electrolytes
RESPIRATORY ACIDOSIS
Definition
Primary ↑PaCO2 (hypercapnia) due to alveolar hypoventilation
Causes
- CNS depression: Opioids, sedatives, benzodiazepines, anesthetics, head trauma, intracranial tumors, stroke, obesity-hypoventilation syndrome (OHS), primary alveolar hypoventilation
- Neuromuscular: ALS, Guillain-Barré, myasthenia gravis, muscular dystrophies, phrenic nerve injury
- Airway obstruction: COPD (#1 chronic), severe asthma, OSA, foreign body
- Pulmonary parenchyma: Severe pneumonia, ARDS, pulmonary fibrosis
- Chest wall: Kyphoscoliosis, flail chest, morbid obesity
- Mechanical ventilation: Improper settings (low respiratory rate/TV); auto-PEEP
Clinical Features
- Acute hypercapnia: Anxiety, dyspnea, confusion, psychosis, hallucinations → coma; CO2 narcosis
- Chronic hypercapnia: Sleep disorders, memory loss, daytime somnolence, personality changes, tremor, myoclonic jerks, asterixis
- "CO2 narcosis" signs: Papilledema, focal weakness, headache, abnormal reflexes (mimics raised ICP)
Compensation
- Acute: HCO3⁻ rises ~1 mEq/L per 10 mmHg ↑PaCO2
- Chronic (days): HCO3⁻ rises ~3.5 mEq/L per 10 mmHg ↑PaCO2
Treatment
- Correct underlying cause
- Non-invasive ventilation (NIV/BiPAP): COPD exacerbation, OHS, neuromuscular disease
- Intubation and mechanical ventilation: If NIV fails or contraindicated
- Avoid rapid correction of chronic respiratory acidosis → metabolic alkalosis ("post-hypercapnic alkalosis") if HCO3⁻ remains elevated after PaCO2 is lowered
RESPIRATORY ALKALOSIS
Definition
Primary ↓PaCO2 (hypocapnia) due to alveolar hyperventilation
Causes
CNS stimulation:
- Pain, anxiety/panic disorder, fever, CVA (especially pontine lesions), meningitis/encephalitis, head trauma, tumor
Hypoxemia/Tissue Hypoxia:
- High-altitude acclimatization (#1 cause in altitude)
- Pneumonia, pulmonary edema, aspiration, PE, severe anemia
Drugs/Hormones:
- Pregnancy (progesterone stimulates respiratory center) → chronic respiratory alkalosis of pregnancy
- Salicylates (early) - direct CNS stimulation
- Cardiac failure
Stimulation of Chest Receptors:
- Hemothorax, flail chest, cardiac failure, pulmonary embolism (important cause)
Miscellaneous:
- Septicemia (gram-negative bacteria/LPS stimulate CNS chemoreceptors) - respiratory alkalosis is the earliest acid-base sign of sepsis
- Hepatic failure (ammonia + progesterone levels stimulate breathing)
- Mechanical hyperventilation
Clinical Features
- Acute hypocapnia: paresthesias (perioral, extremities), lightheadedness, tetany (due to decreased ionized calcium as alkalosis increases protein-Ca binding), carpopedal spasm
- Chronic: usually asymptomatic; compensated by renal HCO3⁻ excretion
- ECG: may show ST changes and arrhythmias
- Importantly: chronic respiratory alkalosis is the one disorder where compensation can fully normalize pH
Compensation
- Acute: HCO3⁻ falls ~2 mEq/L per 10 mmHg ↓PaCO2
- Chronic: HCO3⁻ falls ~5 mEq/L per 10 mmHg ↓PaCO2
Treatment
- Treat underlying cause
- Anxiety/hyperventilation: reassurance, breathing into paper bag, anxiolytics
- Hepatic failure, sepsis: treat underlying disease
MIXED ACID-BASE DISORDERS
Common Clinically Important Mixed Disorders
| Pattern | Example Scenarios | Key Clue |
|---|---|---|
| High-AG MA + Respiratory Alkalosis | Salicylate toxicity, sepsis, hepatic failure with lactic acidosis | pH may be near normal; AG elevated; PaCO2 lower than expected by Winter's |
| High-AG MA + Metabolic Alkalosis | Vomiting + DKA; vomiting + renal failure | Delta ratio >2; HCO3⁻ higher than expected for degree of AG elevation |
| Metabolic Alkalosis + Respiratory Acidosis | COPD patient on diuretics; post-hypercapnic alkalosis | pH near normal; both HCO3⁻ and PaCO2 elevated |
| High-AG MA + Non-AG MA | DKA treated with saline; diarrhea + uremia | Delta ratio <1; more HCO3⁻ drop than AG rise |
| Respiratory Alkalosis + Metabolic Alkalosis | Pregnant patient vomiting; liver failure + NG suction | Very high pH; both HCO3⁻ elevated and PaCO2 depressed |
EXAM PEARL SUMMARY TABLE
| Finding | Most Likely Diagnosis |
|---|---|
| High AG, normal osmolar gap | DKA, lactic acidosis, uremia, salicylates (late), propylene glycol |
| High AG + HIGH osmolar gap | Methanol, ethylene glycol, isopropyl alcohol (early) |
| High AG + normal osmolar gap + no ketones/lactate/uremia | Think 5-oxoproline (acetaminophen) |
| Non-AG acidosis + negative UAG | GI loss (diarrhea) |
| Non-AG acidosis + positive UAG + hypokalemia + urine pH >5.5 | Type 1 distal RTA |
| Non-AG acidosis + positive UAG + hypokalemia + urine pH <5.5 | Type 2 proximal RTA |
| Non-AG acidosis + positive UAG + HYPERkalemia | Type 4 RTA (hyporeninemic hypoaldosteronism) |
| Metabolic alkalosis + low urine Cl⁻ | Saline-responsive (vomiting, remote diuretic) |
| Metabolic alkalosis + high urine Cl⁻ + hypertension | Mineralocorticoid excess (Conn's, Cushing's) |
| Metabolic alkalosis + high urine Cl⁻ + normotension + hypokalemia | Bartter's or Gitelman's syndrome |
| Hypercalcemia + PTH elevated | Primary HPT or FHH |
| Hypercalcemia + PTH suppressed + PTHrP elevated | Malignancy (humoral) |
| Hypercalcemia + PTH suppressed + 1,25(OH)2D elevated | Granulomatous disease / Lymphoma |
| Hypocalcemia + PTH elevated | Vitamin D deficiency, PTH resistance (PHP) |
| Hypocalcemia + PTH low + Mg low | Fix magnesium first |
| Prolonged QT | Hypocalcemia |
| Shortened QT | Hypercalcemia |
| Pelvic pain + bladder filling + negative cultures | IC/BPS - do NOT treat as UTI |
| RBC casts in urine | Glomerulonephritis until proven otherwise |
| UACR >30 mg/g | Initiate workup for kidney disease |
Sources: Harrison's Principles of Internal Medicine, 22nd Edition (2025, McGraw-Hill Medical), Chapters 54-58; Section 7: Alterations in Renal and Urinary Tract Function.
This covers everything in those chapters - from first principles through clinical presentation, diagnosis, and management - at the level Harrison's expects of the best physicians. Master the systematic approach to acid-base (always correct the AG for albumin, always check the delta-delta, always use urine Cl⁻ in alkalosis), and remember that IC/BPS and glomerular disease are frequently misdiagnosed conditions where pattern recognition prevents years of patient suffering.
This is a shared conversation. Sign in to Orris to start your own chat.