Pathogenesis of hep a virus
Reading File
Finding Sources
Searching PubMed
"hepatitis A virus" AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, authoritative information from Harrison's, Goldman-Cecil, Sleisenger & Fordtran's, and Sherris & Ryan's Medical Microbiology. Let me compile the full answer.
Pathogenesis of Hepatitis A Virus (HAV)
1. The Virus - Structure
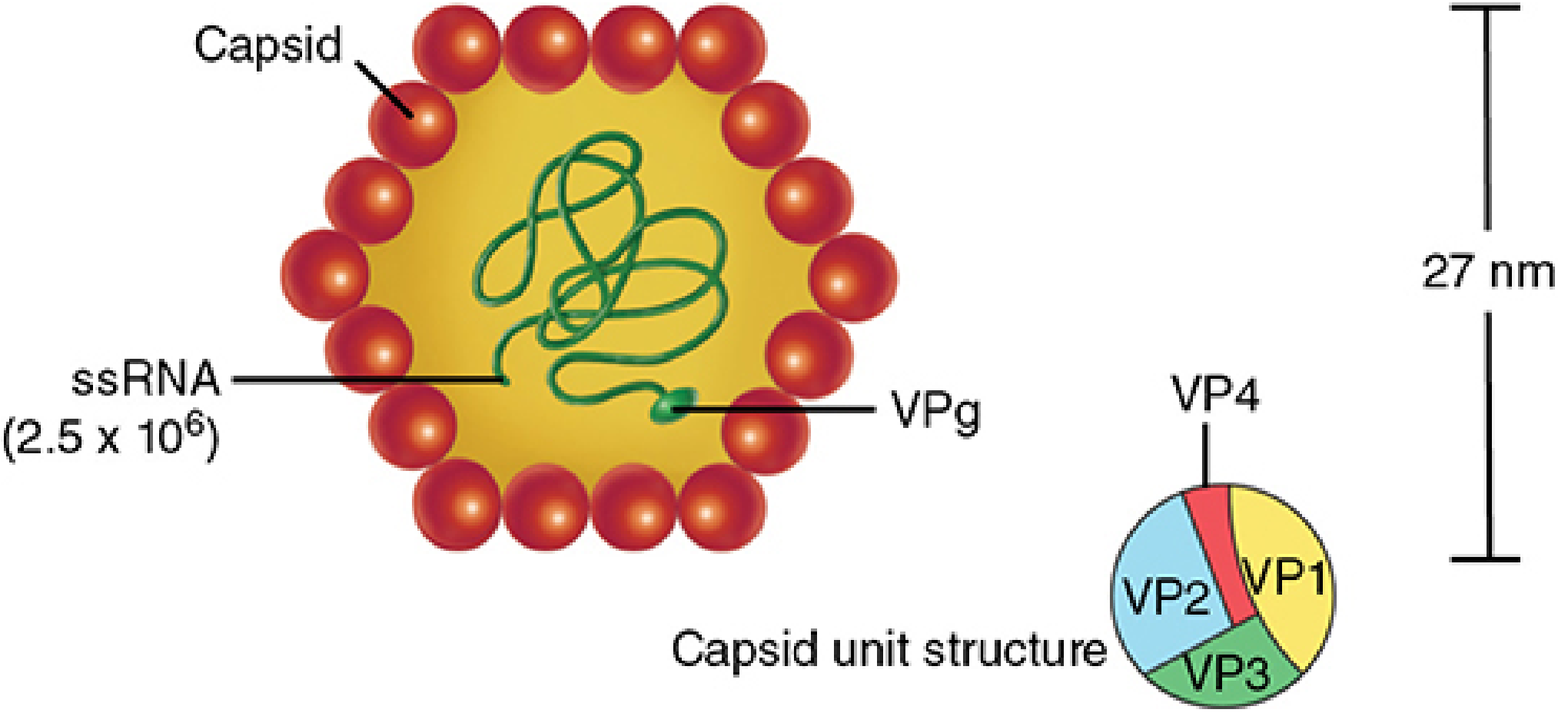
HAV is a 27-28 nm, non-enveloped, icosahedral virus in the family Picornaviridae, genus Hepatovirus. Its genome is a 7.48 kb positive-sense, single-stranded RNA with a VPg protein covalently linked to its 5' end. The capsid has four structural proteins: VP1, VP2, VP3, and VP4. VP1 is the spike protein that binds host cell receptors. Only one serotype exists (multiple genotypes: I, II, III - genotype I predominates globally).
HAV is unusually stable: survives in seawater, dried feces, and acidic environments, but is inactivated by heating to 85°C for 1 minute.
- Sherris & Ryan's Medical Microbiology, p. 480-481
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease, p. 1210
2. Transmission
HAV spreads via the fecal-oral route - either person-to-person or through ingestion of fecally contaminated food or water (shellfish, raw vegetables, drinking water). Blood-borne transmission is rare. High-risk groups include travelers to endemic areas, day care contacts, MSM, injection drug users, and homeless persons.
3. Entry and Intracellular Replication
The steps of the HAV life cycle:
-
Attachment: HAV binds to specific surface receptors on hepatocytes. The primary cellular receptor is TIM-1 (T-cell immunoglobulin and mucin domain 1, also called HAVcr-1), a glycoprotein. Entry occurs via receptor-mediated endocytosis (viropexis).
-
Uncoating: The viral capsid is uncoated inside the cell.
-
Translation: Since HAV RNA is positive-sense, it acts directly as mRNA. Ribosomes bind to the internal ribosomal entry site (IRES) in a cap-independent manner, translating the single open reading frame into a large polyprotein, which is then proteolytically cleaved into mature structural and non-structural proteins.
-
Replication: The non-structural protein RNA-dependent RNA polymerase (RdRp) creates a negative-sense RNA intermediate, which serves as the template for new positive-sense genomic RNA. All replication occurs exclusively in the cytoplasm within a membrane-bound replication complex.
-
Assembly and Release: New viral genomes are packaged into newly synthesized capsid proteins. Virions are exported - primarily into bile (and to a lesser extent into serum). Cell lysis also releases virions. HAV is also known to exist in a quasi-enveloped form (eHAV) wrapped in host-derived membranes during bloodstream transit, which helps it evade immune detection.
- Goldman-Cecil Medicine, p. (block 18)
- Sherris & Ryan's Medical Microbiology, p. 481
4. Spread to the Liver and Viral Shedding
After ingestion, HAV first replicates in the intestinal mucosa during the incubation period (15-45 days). Viremia follows, allowing the virus to reach the liver. Hepatocytes are the primary target. Fecal shedding is maximal during the late incubation period, before clinical symptoms appear - this is why HAV spreads efficiently from asymptomatic individuals.
5. Mechanism of Liver Injury - Immunopathogenesis
This is the central concept: HAV is not directly cytopathic to hepatocytes under ordinary circumstances. The liver damage is primarily immune-mediated.
Key evidence and mechanisms (from Harrison's, 22nd ed.):
-
Viral shedding and peak viremia pre-date clinical liver injury, meaning viral replication and cell death are temporally dissociated.
-
Cytotoxic CD8+ T-cell response is the primary instrument of hepatocyte destruction. HAV-specific CD8+ CTLs recognize viral peptides presented on MHC class I and kill infected hepatocytes.
-
CD4+ helper T cells and IFN-γ secreting CD4+ cells amplify the response.
-
Notably, nonspecific (bystander) CD8+ T-cell activation against non-HAV viral antigens has been demonstrated during acute HAV infection, and this correlates with the degree of liver injury - highlighting that the immune response is broader than just HAV-specific.
-
An activated innate immune response is also evident, suggesting multiple immunologic pathways contribute simultaneously.
-
HAV does not establish chronic infection, and individuals who lack robust cellular immunity (e.g., immunocompromised patients) may have less liver damage but prolonged viremia.
-
Harrison's Principles of Internal Medicine 22E, p. 2693-2694
6. Histopathology of the Infected Liver
The typical histologic lesion seen in acute HAV infection includes:
| Feature | Description |
|---|---|
| Inflammatory infiltrate | Panlobular infiltration with mononuclear cells (primarily small lymphocytes) |
| Hepatocyte injury | Degeneration, necrosis, ballooning, and cell dropout |
| Apoptotic bodies | Councilman (acidophilic) bodies - apoptotic hepatocytes |
| Kupffer cells | Hyperplasia (reactive proliferation) |
| Cholestasis | Variable |
| Reticulin framework | Preserved in uncomplicated cases |
| Regeneration | Mitotic figures, multinucleated cells, rosette formation |
In severe cases, massive hepatic necrosis leads to fulminant hepatic failure (rare, <1% of cases).
- Harrison's Principles of Internal Medicine 22E, p. 2694
7. Clinical Course and Outcomes
- Incubation period: 15-45 days
- Up to 50% of adult infections are subclinical
- Symptomatic illness: fever, anorexia, nausea, right upper quadrant pain, followed by jaundice with dark urine and clay-colored stools
- Self-limiting in the vast majority; full recovery in weeks to months
- No chronic carrier state - HAV does not integrate into the genome or persist
- Relapsing cholestatic hepatitis: HAV is the most common cause (Sleisenger & Fordtran's)
- Fulminant hepatitis with extensive liver necrosis: rare but potentially fatal, more common in elderly and those with pre-existing liver disease
- Rare extrahepatic manifestations: immune complex disorders have been linked (rarely) with HAV
8. Immunity After Infection
After acute infection, IgM anti-HAV appears first (used for diagnosis of acute infection), followed by IgG anti-HAV, which persists lifelong and confers complete protection against reinfection.
Summary Flowchart
Ingestion of contaminated food/water
↓
Replication in intestinal mucosa (incubation 15-45 days)
↓
Viremia → Spread to liver via bloodstream
↓
Receptor-mediated entry into hepatocytes (TIM-1/HAVcr-1)
↓
Cytoplasmic replication (IRES-mediated polyprotein → RdRp)
↓
Viral export into bile → fecal shedding (peak: pre-jaundice)
↓
Host immune recognition (innate → CD4+ & CD8+ T cells)
↓
CD8+ CTL-mediated hepatocyte killing + bystander activation
↓
Liver injury (↑ ALT/AST, jaundice)
↓
Clearance (IgM → IgG), self-limited recovery
↓ (rarely)
Fulminant hepatic failure
Sources: Harrison's Principles of Internal Medicine 22E | Goldman-Cecil Medicine | Sleisenger & Fordtran's Gastrointestinal and Liver Disease | Sherris & Ryan's Medical Microbiology, 8th Ed.
A 2023 review in Nature Reviews Disease Primers (Van Damme, Pintó, Feng) provides the most current synthesis of HAV infection biology, consistent with the above. A 2021 review specifically on HAV pathogenicity and virulence (Pintó et al., Virulence) further details the molecular mechanisms of immune evasion and quasi-enveloped HAV forms.
This is a shared conversation. Sign in to Orris to start your own chat.