Reading File
Running Command
python3 -c " import pdfplumber pdf_path = '/home/daytona/workspace/attachments/…
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a comprehensive answer. Here is the full lecture case analysis:
Diabetes Mellitus - IMD 33rd Batch Lecture Case Analysis
Case Summary: 53-year-old female with 5-year history of Type 2 DM on Gliclazide 60 mg OD + Metformin 500 mg TID, presenting with blurring of vision and dizziness. FBS 220 mg/dL, HbA1c 10%. Admitted and started on insulin therapy.
Objective 1: Physiologic Mechanisms Involved in Diabetes Mellitus
The Endocrine Pancreas
The endocrine pancreas consists of about 1 million clusters called the islets of Langerhans, which contain four major cell types:
- β cells - produce insulin (lower blood glucose)
- α cells - secrete glucagon (raises blood glucose via hepatic glycogenolysis)
- δ cells - secrete somatostatin (suppresses both insulin and glucagon)
- PP cells - secrete pancreatic polypeptide (gastrointestinal effects)
(Robbins, Cotran & Kumar, p. 1014)
Normal Glucose Homeostasis
Glucose homeostasis is tightly regulated by three processes:
- Glucose production in the liver
- Glucose uptake and utilization by peripheral tissues (mainly skeletal muscle)
- Actions of insulin and counter-regulatory hormones (glucagon, cortisol, catecholamines)
During fasting: low insulin + high glucagon → hepatic gluconeogenesis and glycogenolysis → prevents hypoglycemia.
After a meal: insulin rises, glucagon falls → promotes glucose uptake and utilization.
(Robbins, Cotran & Kumar, p. 1015)
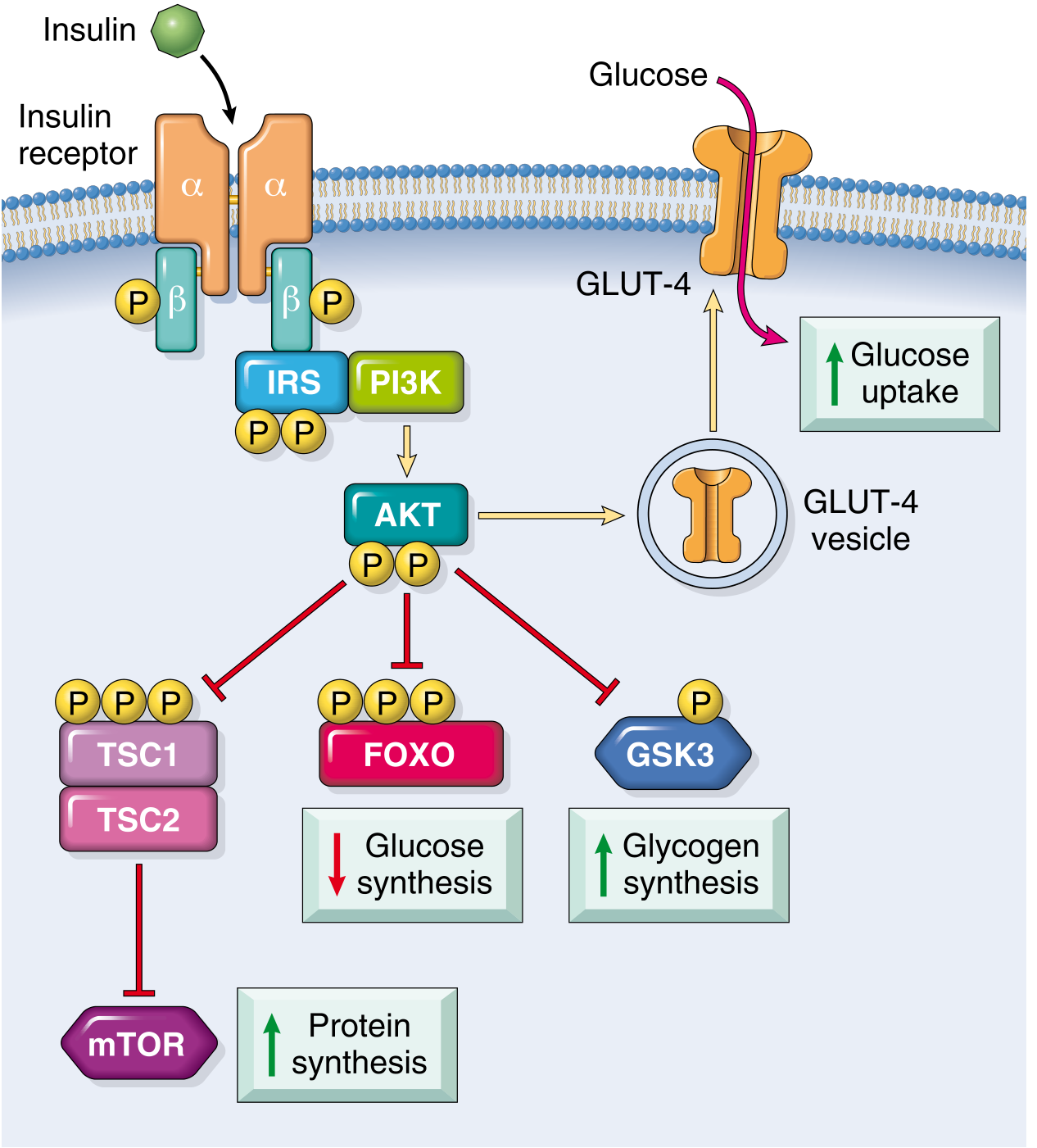
How Insulin Works (Signal Cascade)
The insulin receptor is a tetrameric protein (2α + 2β subunits). Insulin binding to the α-subunit activates β-subunit tyrosine kinase activity, which phosphorylates insulin receptor substrate (IRS) proteins. This activates PI3-kinase → AKT, which:
- Translocates GLUT-4 vesicles to the cell membrane → increased glucose uptake in muscle and fat
- Inhibits FOXO → reduces hepatic gluconeogenesis
- Inhibits GSK-3 → increases glycogen synthesis
- Activates mTOR via TSC1/2 → promotes protein synthesis
(Robbins, Cotran & Kumar, p. 1019)
Objective 2: Physiologic Process Impaired in Type 2 DM
Dual Defect: Insulin Resistance + β-Cell Failure
Type 2 DM (T2DM) accounts for about 90-95% of all DM cases. It is caused by:
- Peripheral insulin resistance - target tissues (muscle, liver, adipose) fail to respond normally to insulin
- Relative insulin deficiency - the pancreatic β-cell secretory response is inadequate to overcome resistance
(Robbins, Cotran & Kumar, p. 1015)
How Insulin Resistance Develops (T2DM Progression)
The three key mediators from excess adipose tissue are: adipokines, free fatty acids (FFAs), and inflammation.
- FFAs: Excess FFAs from central adiposity overwhelm fatty acid oxidation pathways, accumulating toxic lipid metabolites (diacylglycerol, ceramides) that attenuate insulin receptor signaling. In the liver, this allows unchecked gluconeogenesis.
- Adipokines: In obesity, adiponectin (which normally increases insulin sensitivity) is reduced, worsening resistance.
- Inflammation: Proinflammatory cytokines (especially IL-1β via inflammasome activation) cause both insulin resistance and β-cell dysfunction.
(Robbins, Cotran & Kumar, p. 1020)
The Progression to Frank Diabetes
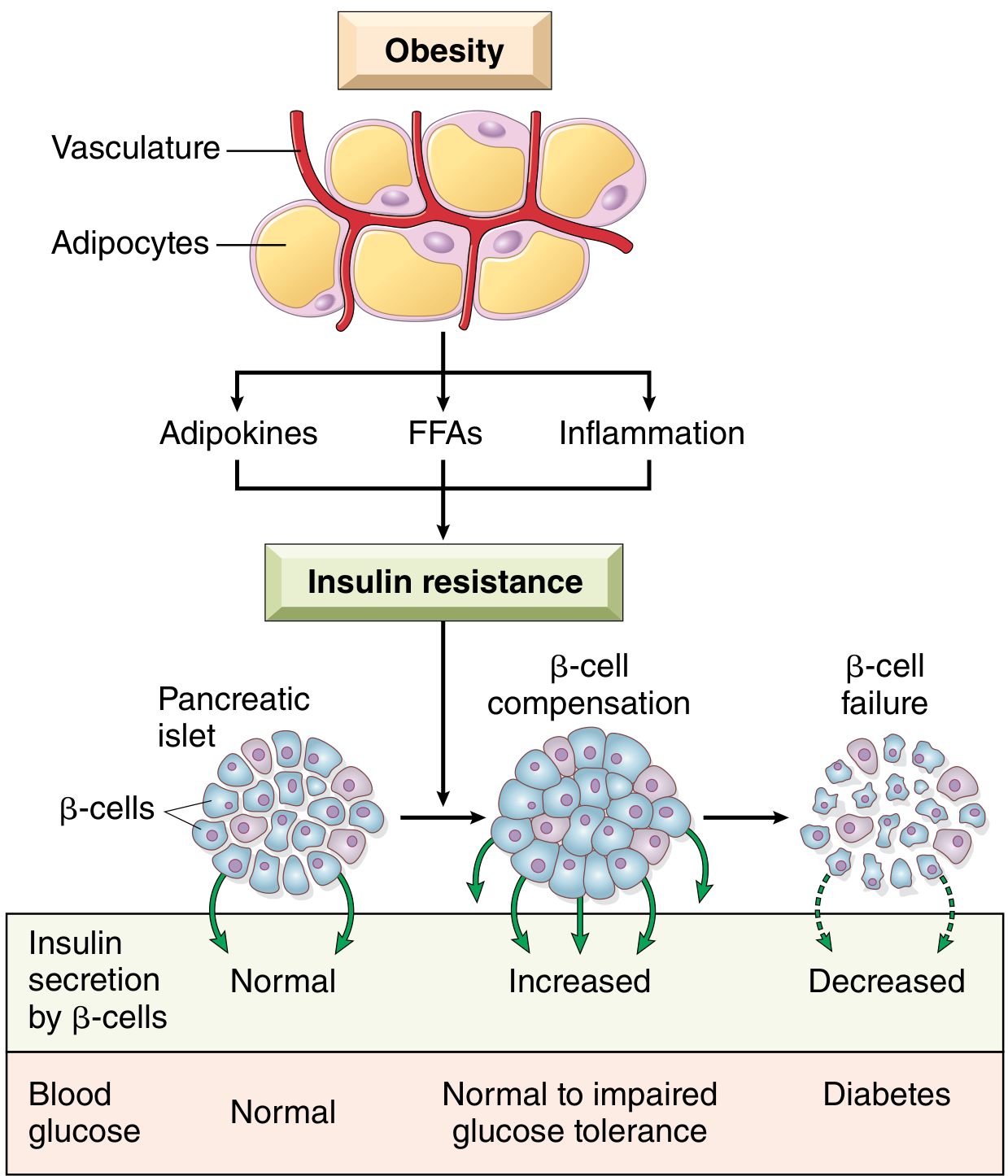
- Initially, β-cells compensate by secreting more insulin (hyperinsulinemia) - blood glucose remains normal to near-normal.
- With prolonged insulin resistance, β-cells fail and cannot secrete enough insulin even under maximal stimulation.
- Moderate hyperglycemia first appears after carbohydrate meals (postprandial), then fasting hyperglycemia develops.
(Guyton and Hall Medical Physiology, p. 974)
Objective 3: Mechanisms of Clinical Manifestations and Laboratory Findings
This Patient's Findings Explained
| Finding | Mechanism |
|---|---|
| FBS 220 mg/dL | Insufficient insulin action → hepatic glucose output unrestrained + reduced peripheral glucose uptake (GLUT-4 not translocated) |
| HbA1c 10% | Nonenzymatic glycation of hemoglobin; reflects average blood glucose over ~120 days; >6.5% = DM; 10% indicates poor chronic control |
| Blurring of vision | Osmotic changes in lens (sorbitol accumulation via polyol pathway) + early diabetic retinopathy from microvascular damage |
| Dizziness | Possibly postural hypotension from autonomic neuropathy or hypoglycemic episodes from current drug therapy |
| Thin/ill-appearing | Protein catabolism - failure to use glucose → increased protein degradation → weight loss and asthenia |
Mechanisms of Chronic Hyperglycemia Causing Organ Damage
Four main mechanisms drive long-term complications ("glucotoxicity"):
-
Advanced Glycation End Products (AGEs): Nonenzymatic reactions of glucose metabolites with amino groups of proteins. AGEs cross-link collagen in vessel walls, trap LDL, and activate macrophages → inflammation and vascular stiffening.
-
Polyol Pathway: Glucose is converted to sorbitol by aldose reductase. Sorbitol accumulates inside cells (especially lens, retina, peripheral nerves, kidneys) causing osmotic injury. Sorbitol is also oxidized to fructose, increasing intracellular oxidative stress.
-
Protein Kinase C (PKC) Activation: Excess intracellular glucose increases diacylglycerol synthesis → activates PKC → abnormal regulation of vascular permeability and blood flow.
-
Hexosamine Pathway: Excess glucose shunted into hexosamine pathway → excess glucosamine formation → altered gene expression and cell function.
(Robbins, Cotran & Kumar, p. 1021)
Diagnostic Criteria (ADA/WHO) - This Patient's Interpretation
| Criterion | Threshold | This Patient |
|---|---|---|
| Fasting plasma glucose | ≥126 mg/dL | 220 mg/dL - well above |
| Random plasma glucose | ≥200 mg/dL | - |
| HbA1c | ≥6.5% | 10% - well above |
| 2-hour OGTT | ≥200 mg/dL | - |
Blood glucose is normally 70-120 mg/dL. This patient's FBS of 220 mg/dL and HbA1c of 10% confirm poorly controlled T2DM. The target HbA1c in most patients is below 7%.
(Robbins, Cotran & Kumar, p. 1014; Lippincott Biochemistry, p. 211)
Objective 4: Physiologic Basis of Management
1. Gliclazide (Sulfonylurea - 2nd Generation)
Mechanism: Gliclazide binds to a 140-kDa high-affinity sulfonylurea receptor (SUR1) associated with the ATP-sensitive potassium channel (K-ATP channel) on β-cells. This:
- Blocks K+ efflux → membrane depolarization
- Opens voltage-gated Ca²+ channels → calcium influx
- Triggers exocytosis of insulin-containing granules → enhanced insulin secretion
Gliclazide is a 2nd-generation sulfonylurea with greater receptor affinity and fewer drug interactions than 1st-generation agents. It acts regardless of blood glucose level - hence the risk of hypoglycemia (which may explain her dizziness).
(Katzung's Basic and Clinical Pharmacology 16e, p. 1184)
2. Metformin (Biguanide)
Mechanism: Metformin primarily acts by suppressing hepatic glucose production (gluconeogenesis). It does this by activating AMPK (AMP-activated protein kinase), which inhibits gluconeogenic enzymes. Additional effects:
- Increases peripheral tissue sensitivity to insulin
- Reduces intestinal glucose absorption
- Does NOT cause hypoglycemia alone (no effect on insulin secretion directly)
- Promotes modest weight loss - beneficial in overweight patients
(Guyton and Hall Medical Physiology, p. 975)
3. Why Insulin Was Added
This patient despite dual oral therapy (sulfonylurea + metformin) still has:
- FBS 220 mg/dL (target <130 mg/dL)
- HbA1c 10% (target <7%)
This indicates secondary failure of oral agents - the β-cells have likely become sufficiently dysfunctional that adequate insulin levels cannot be maintained. In later stages of T2DM, insulin administration is usually required to control plasma glucose levels.
(Guyton and Hall Medical Physiology, p. 975)
Insulin therapy acts by:
- Directly replacing the deficient/insufficient endogenous insulin
- Restoring GLUT-4 translocation in muscle and fat → normalizing glucose uptake
- Suppressing hepatic gluconeogenesis
- Promoting glycogen synthesis and protein anabolism
Different insulin formulations vary by duration: "Regular" insulin lasts 3-8 hours, while longer-acting forms (zinc/protein precipitates) last 10-48 hours. A typical regimen combines long-acting basal insulin with meal-time short-acting doses.
(Guyton and Hall Medical Physiology, p. 974)
Summary of Management Hierarchy in T2DM
| Step | Intervention | Physiologic Basis |
|---|---|---|
| 1 | Lifestyle: caloric restriction, exercise, weight loss | Reduces adiposity → decreases FFA-driven insulin resistance |
| 2 | Metformin | Suppresses hepatic gluconeogenesis; AMPK activation |
| 3 | Add sulfonylurea (Gliclazide) | Closes K-ATP channel → β-cell depolarization → insulin release |
| 4 | Insulin therapy | Direct replacement; overcomes β-cell failure |
Key Takeaways for This Case
- This 53-year-old woman represents the classic natural history of T2DM: insulin resistance (central adiposity pathway) → β-cell compensation → β-cell exhaustion → overt hyperglycemia.
- Her FBS of 220 mg/dL and HbA1c of 10% indicate both acute and chronic poor glycemic control.
- Blurring of vision is an early sign of diabetic retinopathy via AGE formation and polyol pathway in retinal microvasculature.
- Dizziness could reflect hypoglycemia (sulfonylurea effect) or early autonomic neuropathy.
- The switch to insulin is physiologically appropriate given failure of dual oral therapy - direct exogenous insulin bypasses the failed β-cell secretion.
Primary sources: Guyton and Hall Textbook of Medical Physiology 15th Ed., Ch. 79; Robbins, Cotran & Kumar Pathologic Basis of Disease (2023); Katzung's Basic and Clinical Pharmacology 16th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.