I now have rich, authoritative data from multiple textbooks. Let me compose the complete, comprehensive learning note.
COMPLETE LEARNING NOTE: NSAIDs (Non-Steroidal Anti-Inflammatory Drugs)
From Absolute Beginner to MBBS Examination Mastery
SECTION 1: BIG PICTURE OVERVIEW
"What Problem Does This Drug Class Solve?"
Imagine you accidentally hit your thumb with a hammer. Within minutes, your thumb turns red, swells up, feels hot, and hurts. You cannot use it comfortably. This is inflammation - your body's emergency repair system switching on.
Inflammation is not always your enemy. In fact, it is often your friend. It brings immune cells to fight infection and starts the healing process. BUT - sometimes inflammation becomes excessive, prolonged, and damaging. In diseases like arthritis, it keeps going for years, destroying the very joints it is supposed to protect. In fever, it makes the brain overheat. In period pain, it causes the uterus to cramp. In headaches and post-surgical pain, it creates unbearable suffering.
The central problem: Inflammation produces chemicals called prostaglandins (think of them as little chemical alarm signals). These prostaglandins cause:
- Pain - by sensitizing nerve endings so that even light touch hurts
- Fever - by resetting the brain's thermostat to a higher temperature
- Swelling - by making blood vessels leaky so fluid pours into tissue
- Redness and heat - by increasing blood flow to the area
What NSAIDs do: They block the production of prostaglandins at their source, like shutting off the alarm system factory. Less prostaglandin = less pain, less fever, less swelling.
The target of NSAIDs: An enzyme called cyclooxygenase (COX) - the key enzyme that makes prostaglandins. No COX activity = no prostaglandins = relief.
In one sentence: NSAIDs are drugs that relieve pain, reduce fever, and decrease inflammation by blocking the enzyme (COX) that produces the chemical alarms (prostaglandins) of the body.
SECTION 2: BUILD THE FOUNDATION
2A. Normal Physiology - The Arachidonic Acid Pathway
Before understanding NSAIDs, you need to understand what happens normally inside your cells when injury or inflammation occurs.
Step 1: Membrane Phospholipids
Every cell in your body is surrounded by a membrane (like the skin of a soap bubble). This membrane contains fat-like molecules called phospholipids. Tucked inside these phospholipids is a fatty acid called arachidonic acid (AA).
Think of arachidonic acid as a sleeping soldier inside the membrane, waiting for an emergency signal.
Step 2: The Emergency Signal - Phospholipase A2
When a cell is damaged, or when cytokines (immune signaling molecules) arrive, an enzyme called phospholipase A2 wakes up. It cuts arachidonic acid free from the membrane.
Think of phospholipase A2 as the officer who shouts "Wake up soldiers! We have an emergency!"
Step 3: The Fork in the Road - Two Paths for Arachidonic Acid
Once free, arachidonic acid can take one of two roads:
ARACHIDONIC ACID (freed from membrane)
|
_____|_____
| |
COX 5-Lipoxygenase
PATH PATH
| |
Prostaglandins Leukotrienes
Thromboxane A2 (cause bronchoconstriction
Prostacyclin and allergic reactions)
|
These cause:
PAIN, FEVER, INFLAMMATION,
platelet aggregation, gastric protection
NSAIDs block the COX path. They do NOT block the lipoxygenase path. This is why NSAIDs reduce pain and fever but do NOT treat asthma (which requires leukotriene blockers like montelukast).
Step 4: The COX Enzyme - Two Forms
This is where it gets really important. There are two main forms of the COX enzyme:
| Feature | COX-1 | COX-2 |
|---|
| Expression | Present all the time ("constitutive") in most cells | Switched on ("induced") by inflammation, injury, cytokines |
| Location | Stomach lining, platelets, kidneys, blood vessels | Inflammatory cells, kidney, brain, endothelium |
| Main products | Gastric protective prostaglandins (PGE2, PGI2), thromboxane A2 in platelets | PGE2 and PGI2 at sites of inflammation |
| Main function | Housekeeper - protects stomach, helps platelets clump, maintains kidney blood flow | Emergency worker - drives inflammation, pain, fever |
| Analogy | The maintenance crew keeping the building running | The fire brigade called in when there is a fire |
This distinction is the most important thing to understand about NSAIDs. When you give a drug that blocks BOTH COX-1 and COX-2 (a traditional/non-selective NSAID like ibuprofen), you get:
- Desired: Less inflammation, less pain, less fever (COX-2 block)
- Unwanted: Stomach ulcers (COX-1 block in stomach), bleeding tendency (COX-1 block in platelets), kidney problems (COX-1+2 block in kidney)
2B. Prostaglandins and Their Actions - What They Do
Prostaglandins (PGs) are local signaling molecules - they act close to where they are made, not at distant sites like hormones. The major ones:
| Prostaglandin | Made Where | Key Effects |
|---|
| PGE2 | Widely (inflammatory cells, stomach, kidney, brain) | Pain sensitization, fever, gastric protection, vasodilation, uterine contraction |
| PGI2 (Prostacyclin) | Vascular endothelium | Vasodilation, prevents platelet clumping, protects kidneys |
| TxA2 (Thromboxane A2) | Platelets | Powerful platelet aggregation, vasoconstriction |
| PGD2 | Mast cells | Allergic inflammation, bronchoconstriction |
| PGF2α | Uterus, lungs | Uterine contraction, bronchoconstriction |
Key insight for NSAID pharmacology:
- COX-1 in platelets → TxA2 → platelet aggregation. Aspirin IRREVERSIBLY blocks this → anti-platelet effect lasts 7-10 days (platelet lifetime)
- COX-2 in endothelium → PGI2 → prevents platelet clumping and dilates vessels. Selective COX-2 inhibitors block this without blocking TxA2 → NET EFFECT: THROMBOSIS RISK
2C. How Prostaglandins Cause Pain
Pain nerve endings (nociceptors) normally need a fairly strong stimulus to fire. PGE2 and PGI2 lower the threshold of these nociceptors - they become hypersensitive. Even light touch, normal pressure, or mild temperature changes now cause pain. This is called peripheral sensitization.
Think of it this way: normally, a door needs a hard push to open. PGE2 oils the hinges so the door swings open with the lightest touch.
Centrally, PGE2 in the spinal cord causes central sensitization - the spinal cord neurons become hyperexcitable, amplifying pain signals from the periphery. NSAIDs reduce both peripheral and central sensitization.
2D. How Prostaglandins Cause Fever
The brain has a temperature control center in the hypothalamus (think of it as a thermostat). Normally it is set at 37°C. When infection or inflammation occurs:
- Immune cells release cytokines - IL-1β, IL-6, TNF-α (messengers that say "emergency!")
- Cytokines act on the hypothalamus, inducing COX-2
- COX-2 makes PGE2 in the hypothalamus
- PGE2 resets the thermostat to 39°C or higher
- Body generates more heat and reduces heat loss
- Result: Fever
NSAIDs block COX-2 in the hypothalamus → block PGE2 production → thermostat resets to normal → fever breaks.
Note: Paracetamol (acetaminophen) also works centrally but has no peripheral anti-inflammatory effect.
2E. How Prostaglandins Protect the Stomach
This is critical for understanding NSAID side effects.
The stomach normally digests your food with strong acid. It needs to protect itself from being digested too. It does this by:
- Making a thick layer of mucus (physical barrier)
- Secreting bicarbonate (chemical buffer against acid)
- Maintaining good blood flow to keep cells healthy
- Limiting acid secretion
COX-1 in the stomach lining makes PGE2 and PGI2, which stimulate all of the above protective mechanisms.
Block COX-1 (with non-selective NSAIDs) → Less gastric prostaglandins → Thin mucus, less bicarbonate, poor blood flow, more acid → ULCERS
This is why gastric ulceration is the #1 side effect of non-selective NSAIDs.
SECTION 3: DRUG CLASS FRAMEWORK
3A. Definition of NSAIDs
NSAIDs = Non-Steroidal Anti-Inflammatory Drugs
- "Non-steroidal" = they reduce inflammation WITHOUT using steroids (like cortisone)
- "Anti-inflammatory" = they reduce the inflammatory process
- The term distinguishes them from steroids (like prednisolone), which also reduce inflammation but work differently and have different side effects
They also have:
- Analgesic (pain-relieving) effect
- Antipyretic (fever-reducing) effect
Some, like aspirin, additionally have antiplatelet effect at low doses.
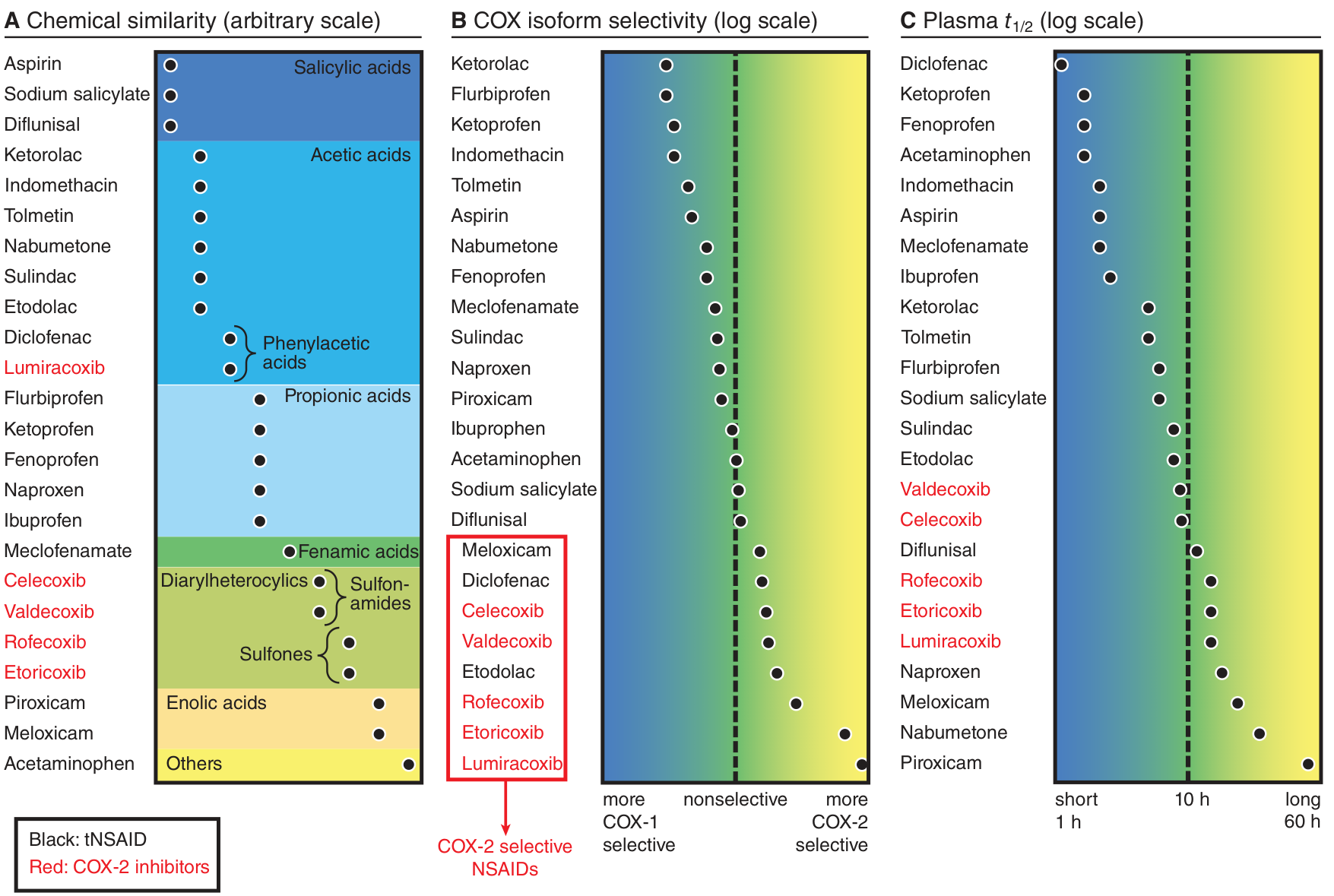
3B. Classification of NSAIDs
By COX Selectivity:
| Class | Examples | COX-1:COX-2 Inhibition |
|---|
| Non-selective (traditional) NSAIDs (tNSAIDs) | Aspirin, ibuprofen, naproxen, indomethacin, diclofenac, ketorolac | Inhibit both COX-1 and COX-2 |
| Preferential COX-2 inhibitors | Meloxicam, nimesulide, diclofenac (higher doses), nabumetone | Slightly prefer COX-2 but still inhibit COX-1 at higher doses |
| Selective COX-2 inhibitors (Coxibs) | Celecoxib, etoricoxib, rofecoxib (withdrawn), valdecoxib (withdrawn) | Selectively inhibit COX-2, spare COX-1 |
By Chemical Structure (important for exams):
| Chemical Group | Examples |
|---|
| Salicylic acids | Aspirin, diflunisal |
| Acetic acids | Indomethacin, ketorolac, diclofenac, etodolac, sulindac, tolmetin, nabumetone |
| Propionic acids | Ibuprofen, naproxen, ketoprofen, flurbiprofen, fenoprofen |
| Fenamic acids (Anthranilic acids) | Mefenamic acid, meclofenamate |
| Enolic acids (Oxicams) | Piroxicam, meloxicam, tenoxicam |
| Diarylheterocyclics | Celecoxib, rofecoxib, valdecoxib, etoricoxib |
| Others | Acetaminophen/Paracetamol (special category) |
(Refer to figure below showing chemical similarity, COX selectivity, and plasma half-life)
Classification of NSAIDs by chemical similarity (A), COX isoform selectivity (B), and plasma t½ (C). - Goodman & Gilman's
3C. Mechanism of Action of NSAIDs
The Core Mechanism (applicable to all NSAIDs):
NSAIDs inhibit the enzyme cyclooxygenase (COX), also called prostaglandin G/H synthase (PGG/H synthase). This enzyme converts arachidonic acid into PGG2 and then PGH2, the unstable intermediates from which all prostaglandins, thromboxane A2, and prostacyclin are eventually made.
No COX → No PGH2 → No downstream prostaglandins, thromboxanes, or prostacyclin
The molecular detail:
- COX enzymes have a hydrophobic channel through which arachidonic acid slides to reach the catalytic site deep inside the enzyme
- Most NSAIDs physically block this channel (reversibly)
- Aspirin is unique - it covalently acetylates a serine residue (Ser-530 in COX-1, Ser-516 in COX-2) inside the channel, permanently destroying enzyme activity
Why COX-2-selective drugs work structurally:
COX-2 has a larger, wider binding channel with an extra side pocket, compared to COX-1. COX-2-selective inhibitors (coxibs) have bulky side groups that fit into this extra pocket in COX-2 but are physically blocked from entering the smaller COX-1 channel. It is like having a key with an extra bump - it only fits the larger lock.
3D. Aspirin - A Drug Worthy of Special Study
Aspirin (acetylsalicylic acid) is the oldest and most unique NSAID. It deserves special attention because it behaves differently from all other NSAIDs.
What makes aspirin unique:
-
Irreversible inhibition - Aspirin acetylates COX enzymes covalently. Once acetylated, the enzyme is permanently destroyed. The cell has to make new enzyme protein (which takes hours to days). Other NSAIDs only reversibly block COX.
-
Dose-dependent effects:
| Dose | Effect | Mechanism |
|---|
| Low (75-325 mg/day) | Antiplatelet (anti-thrombotic) | Irreversibly inhibits COX-1 in platelets → no TxA2 → no platelet aggregation |
| Moderate (325-650 mg every 4-6 hrs) | Analgesic and antipyretic | COX inhibition in peripheral tissues and hypothalamus |
| High (>3-4 g/day) | Anti-inflammatory | Sustained high COX inhibition |
| Very high (toxic) | Salicylism/salicylate toxicity | See adverse effects |
- Platelet effect is permanent for platelet lifetime:
Platelets have no nucleus (they cannot make new protein). When aspirin kills their COX-1, it stays dead for the entire 7-10 day platelet lifespan. This is why aspirin's antiplatelet effect requires only once-daily dosing, and why you must stop aspirin 7-10 days before surgery.
Other NSAIDs (like ibuprofen) also inhibit platelet COX-1, BUT reversibly - so the effect wears off when the drug leaves the system.
Clinical impact: Ibuprofen can compete with aspirin for binding to COX-1. If a patient on low-dose aspirin (for heart protection) takes ibuprofen first, the ibuprofen binds reversibly to COX-1 and physically blocks aspirin from getting in to acetylate it. When ibuprofen leaves, COX-1 is intact again - aspirin's protective effect is undermined. This is a major drug interaction.
3E. Individual NSAIDs - Key Properties
IBUPROFEN
- Propionic acid group
- Short half-life (~2 hours) - needs dosing 3-4 times daily
- One of the safest NSAIDs for GI tract (still causes GI problems but less than indomethacin)
- OTC availability in most countries
- Used for: mild to moderate pain, dysmenorrhea, fever, arthritis
- Can close patent ductus arteriosus (PDA) in premature neonates (COX inhibition reduces PGE2 that keeps ductus open)
- Special: Ibuprofen may cause hypogonadism with prolonged use
NAPROXEN
- Propionic acid group
- Long half-life (~14 hours) - twice daily dosing
- Among the non-selective NSAIDs, naproxen has relatively lower cardiovascular risk compared to ibuprofen or diclofenac
- Used for: musculoskeletal pain, dysmenorrhea, gout, arthritis
INDOMETHACIN
- Acetic acid group
- Very potent COX inhibitor
- Most GI-toxic of common NSAIDs
- High CNS penetration - causes headache, dizziness, confusion (especially in elderly)
- Special uses: acute gout (drug of choice in some guidelines), patent ductus arteriosus closure in neonates (more commonly used than ibuprofen in some centers), ankylosing spondylitis, pericarditis
- High anti-inflammatory potency makes it preferred for acute crystal arthropathies
KETOROLAC
- Acetic acid group
- One of the most potent analgesic NSAIDs
- Available IV/IM - the only parenteral NSAID widely used for short-term pain management
- Maximum 5 days use (high GI/renal toxicity risk with longer use)
- Used for: post-operative pain, renal colic (given IV)
- NOT for chronic use
DICLOFENAC
- Phenylacetic acid group
- Moderate COX-2 preference among traditional NSAIDs (though not truly selective)
- Available oral, topical (gel), IV
- Used for: musculoskeletal pain, arthritis, dysmenorrhea
- Topical diclofenac = effective for local pain with minimal systemic absorption
PIROXICAM
- Enolic acid (Oxicam) group
- Extremely long half-life (45-50 hours) - once daily dosing
- High GI toxicity (long half-life means sustained COX-1 inhibition in stomach)
- Risk of gastric bleeding is higher than shorter-acting NSAIDs
- Used for: arthritis (convenient once-daily)
MELOXICAM
- Enolic acid (Oxicam) group
- Preferential COX-2 inhibitor (not fully selective, but preference for COX-2)
- Better GI tolerability than non-selective NSAIDs
- Once-daily dosing
- Used for: osteoarthritis, rheumatoid arthritis
MEFENAMIC ACID
- Fenamic acid group
- Commonly used for dysmenorrhea (period pain) - dual action: COX inhibition + blocks prostaglandin receptor
- Short-term use only (not beyond 7 days)
- Can cause hemolytic anemia with prolonged use
CELECOXIB
- Diarylheterocyclic (sulfonamide) - COX-2 selective inhibitor (Coxib)
- Spares COX-1 → much lower gastric ulcer risk than non-selective NSAIDs
- Does NOT inhibit platelet aggregation (platelets use COX-1, not COX-2)
- Cardiovascular risk: Inhibits PGI2 (from endothelial COX-2) without inhibiting TxA2 → prothrombotic imbalance → increased risk of MI and stroke
- Contraindicated in sulfonamide allergy (celecoxib has a sulfonamide side chain)
- Used for: osteoarthritis, rheumatoid arthritis, familial adenomatous polyposis (reduces colonic polyps)
- Currently the only commercially available coxib after rofecoxib/valdecoxib withdrawals
ROFECOXIB (withdrawn) and VALDECOXIB (withdrawn)
- Highly selective COX-2 inhibitors
- Withdrawn from market due to significantly increased cardiovascular events (VIGOR trial showed rofecoxib doubled MI risk vs. naproxen)
- Landmark lesson in pharmacovigilance
PARACETAMOL/ACETAMINOPHEN
- Technically not a classic NSAID (no peripheral anti-inflammatory effect at usual doses)
- Acts centrally: inhibits a COX variant in the CNS (possibly COX-3 or the peroxide site of COX enzymes)
- Antipyretic + analgesic - but NO significant anti-inflammatory effect
- No gastric side effects (does not inhibit gastric COX-1)
- No platelet effect
- Key danger: Hepatotoxicity in overdose
- Mechanism of hepatotoxicity: Normal doses are conjugated safely. Overdose overwhelms conjugation, metabolized by CYP2E1 to toxic metabolite NAPQI (N-acetyl-p-benzoquinoneimine). NAPQI depletes glutathione and binds liver proteins → centrilobular necrosis
- Antidote: N-Acetylcysteine (NAC) - replenishes glutathione
3F. Adverse Effects of NSAIDs - Explained Mechanistically
1. GASTROINTESTINAL EFFECTS (Most Common)
Mechanism: NSAIDs reduce gastric mucosal prostaglandins (COX-1 inhibition) + direct mucosal irritation (acidic drug damages mucosa directly).
Less PGE2 and PGI2 in stomach → thinner mucus layer → less bicarbonate → reduced mucosal blood flow → reduced ability to resist acid → erosions, ulcers, bleeding, perforation
Spectrum: Dyspepsia (most common, 10-30%), gastric erosions, peptic ulcer disease, GI bleeding (most dangerous), perforation.
Risk factors for GI toxicity:
- Age >60
- History of peptic ulcer
- H. pylori infection
- Concomitant corticosteroids or anticoagulants
- High dose or long duration NSAID use
- Non-selective NSAIDs (not coxibs)
- Piroxicam has higher GI risk than ibuprofen
Prevention:
- Use COX-2 selective inhibitors (celecoxib)
- Add a proton pump inhibitor (PPI) like omeprazole
- Use the lowest effective dose
- If both GI and CV protection needed: celecoxib + PPI is common strategy
2. CARDIOVASCULAR EFFECTS
Mechanism:
- COX-2 in vascular endothelium makes PGI2 (prostacyclin) → vasodilates and prevents platelet aggregation
- COX-1 in platelets makes TxA2 → promotes platelet aggregation and vasoconstriction
- Normal balance: PGI2 vs TxA2 = pro-thrombotic vs anti-thrombotic balance
Selective COX-2 inhibitors (Coxibs): Block endothelial PGI2 WITHOUT blocking platelet TxA2 → TxA2 wins → pro-thrombotic state → increased MI and stroke risk.
Non-selective NSAIDs: Block both, but in high-risk patients (existing CV disease), the loss of PGI2 still tips the balance.
Naproxen exception: Among non-selective NSAIDs, naproxen appears to have the lowest cardiovascular risk, possibly because its long half-life provides more sustained TxA2 inhibition relative to PGI2.
Current guidelines: NSAIDs and coxibs should be avoided wherever possible in patients with ischemic heart disease, previous thrombosis, or poorly controlled hypertension. When needed, use the lowest dose for the shortest duration.
3. RENAL EFFECTS
Background physiology: Prostaglandins (PGE2 and PGI2) produced in the kidney dilate the afferent arteriole and help maintain glomerular filtration rate (GFR) and renal blood flow. This is especially important when the circulation is stressed (heart failure, dehydration, liver cirrhosis, use of ACE inhibitors or diuretics).
Mechanism of NSAID renal toxicity: In normal people with healthy circulation and good hydration, prostaglandins are not critical for kidney function. But in a person with poor blood flow to the kidney (heart failure, dehydration), prostaglandins are working hard to dilate the kidney's arteries and maintain filtration.
Block COX → Block prostaglandins → Kidney arteries constrict → GFR falls → Acute Kidney Injury (AKI)
Other renal effects:
- Salt and water retention → edema, worsening hypertension, worsening heart failure
- Hyponatremia (rare)
- Hyperkalemia (reduced aldosterone effect due to reduced renin from prostaglandin inhibition)
- Interstitial nephritis (rare, idiosyncratic) - seen especially with fenoprofen, ibuprofen - can be associated with nephrotic syndrome (minimal change disease pattern)
Note: Both COX-1 and COX-2 are expressed in the kidney. Even COX-2-selective inhibitors cause renal adverse effects.
4. PLATELET AND BLEEDING EFFECTS
Mechanism: COX-1 in platelets makes TxA2 → promotes platelet aggregation. NSAIDs block this → impaired platelet aggregation → prolonged bleeding time.
Non-selective NSAIDs: Reversible inhibition - effect lasts as long as drug is in system (usually 4-12 hours depending on drug)
Aspirin: Irreversible - lasts 7-10 days (platelet lifetime)
Coxibs (celecoxib): Do NOT significantly inhibit platelet aggregation (platelets use COX-1, which coxibs spare)
Clinical implications:
- Stop NSAIDs before elective surgery (aspirin: 7-10 days; others: 1-3 half-lives before)
- Avoid NSAIDs in patients on anticoagulants (dual risk: impaired platelet function + GI ulcer that bleeds)
5. RESPIRATORY EFFECTS - ASPIRIN-EXACERBATED RESPIRATORY DISEASE (AERD)
Previously called: Aspirin-Sensitive Asthma or Samter's Triad (Asthma + Nasal polyps + Aspirin sensitivity)
Mechanism (important for exams):
When COX is blocked, arachidonic acid cannot go down the prostaglandin pathway. It is then shunted down the lipoxygenase pathway → more leukotrienes (LTC4, LTD4, LTE4) → bronchoconstriction, bronchospasm
This is why asthmatics can get severe bronchospasm with aspirin or any non-selective NSAID. Patients with nasal polyps are at highest risk.
All NSAIDs (including coxibs) can potentially trigger this - avoid in AERD
6. REYE'S SYNDROME (Aspirin-specific)
Aspirin in children under 12 years with viral infections (especially influenza or chickenpox) → can cause Reye's syndrome = acute non-inflammatory encephalopathy + fatty liver degeneration
Mechanism: Aspirin appears to interfere with mitochondrial function, especially when viral infection has already stressed hepatic mitochondria.
Aspirin is therefore CONTRAINDICATED in children and adolescents with febrile viral illness. Use paracetamol or ibuprofen instead.
7. SALICYLATE TOXICITY (Aspirin overdose)
Stages:
- Early: Nausea, vomiting, tinnitus (ringing in ears), dizziness
- Intermediate: Respiratory alkalosis (aspirin directly stimulates respiratory center → hyperventilation → CO2 blown off)
- Late (severe overdose): Metabolic acidosis (accumulation of organic acids, uncoupling of oxidative phosphorylation) PLUS respiratory alkalosis = mixed acid-base disorder (classic exam finding)
- Terminal: Hyperthermia, convulsions, coma, pulmonary edema
Treatment: Urine alkalinization with sodium bicarbonate (ion trapping - ionized salicylate cannot cross tubular cells back into blood, so gets excreted in urine), dialysis in severe cases.
8. DRUG INTERACTIONS (Key ones)
| NSAID | Drug | Interaction | Mechanism |
|---|
| All NSAIDs | Warfarin | Increased bleeding risk | Displace warfarin from protein binding + GI ulceration |
| All NSAIDs | ACE inhibitors / ARBs | Worsened renal function | Both reduce renal prostaglandins; combined = AKI risk |
| All NSAIDs | Diuretics | Reduced diuretic effect + AKI | NSAIDs retain Na+; reduce renal prostaglandins |
| All NSAIDs | Lithium | Increased lithium toxicity | NSAIDs reduce lithium excretion by kidneys |
| All NSAIDs | Methotrexate | Methotrexate toxicity | NSAIDs reduce renal tubular secretion of methotrexate |
| Ibuprofen | Aspirin (low dose) | Reduced aspirin antiplatelet effect | Ibuprofen blocks aspirin's access to COX-1 |
| Aspirin | NSAIDs | Same as above | |
3G. Contraindications
Absolute:
- Active peptic ulcer disease (non-selective NSAIDs, relative for coxibs with PPI)
- Severe renal failure
- Severe hepatic failure
- Aspirin in children/adolescents with viral illness (Reye's syndrome risk)
- Celecoxib in sulfonamide allergy
- Third trimester of pregnancy (risk of premature closure of ductus arteriosus, delayed labor, oligohydramnios, pulmonary hypertension)
Relative/Use with caution:
- Asthma (risk of AERD) - use with extreme caution
- Heart failure
- Hypertension
- History of GI bleeding or ulcer
- Elderly patients
- Pregnancy (first trimester: limited; second trimester: caution; third trimester: contraindicated)
- Concomitant anticoagulant use
- Renal impairment (even mild)
- Hepatic impairment
3H. Specific Clinical Uses
| Condition | Drug(s) of Choice | Why |
|---|
| Mild-moderate pain | Ibuprofen, naproxen | Effective, relatively safe |
| Dysmenorrhea (period pain) | Mefenamic acid, ibuprofen, naproxen | Prostaglandin drives uterine cramps; COX inhibition relieves |
| Acute gout | Indomethacin, ibuprofen, naproxen | Potent anti-inflammatory; indomethacin fastest onset |
| Osteoarthritis | Ibuprofen, diclofenac, celecoxib (if GI risk) | Reduce inflammation and pain |
| Rheumatoid arthritis (symptomatic) | NSAIDs as adjuncts to DMARDs | Symptom relief; do not modify disease |
| Ankylosing spondylitis | Indomethacin, diclofenac, naproxen | High anti-inflammatory potency needed |
| Patent ductus arteriosus (premature neonates) | Indomethacin, ibuprofen IV | Block PGE2 that keeps ductus open |
| Fever | Ibuprofen, paracetamol | Antipyretic via COX-2 inhibition in hypothalamus |
| Post-operative pain | Ketorolac IV/IM | Parenteral NSAID; opioid-sparing |
| Pericarditis | Ibuprofen, aspirin | Anti-inflammatory |
| Familial adenomatous polyposis (FAP) | Celecoxib | COX-2 overexpressed in colonic polyps; COX-2 inhibition reduces polyp formation |
| Antiplatelet (coronary artery disease, stroke prevention) | Aspirin (low dose: 75-150 mg) | Irreversible COX-1 block → permanent antiplatelet effect |
| Preeclampsia prevention | Aspirin (low dose) | Anti-thrombotic; reduces thromboxane-mediated placental vasoconstriction |
| Migraine (acute) | Ibuprofen, naproxen, aspirin | Prostaglandin-mediated vasodilation and sensitization |
| Renal colic | Ketorolac IV, diclofenac | Reduce ureteral prostaglandin-driven spasm + analgesic |
SECTION 4: TEACH USING ANALOGIES
Analogy 1: The Alarm Factory (Core Mechanism)
Imagine your body has a factory that produces "Pain Alarm" chemicals (prostaglandins). When you are injured, the factory goes into overdrive - churning out alarm after alarm. You feel pain, you burn with fever, you swell up.
NSAIDs are workers who sneak into the factory and break the main machine (COX enzyme). No machine = no alarm chemicals = no pain signals sent.
Non-selective NSAIDs break ALL the machines everywhere - including the machines in the stomach that produce "protective alarm" chemicals (which actually protect the stomach lining). Break those too → stomach unprotected → ulcer.
COX-2 selective NSAIDs are like saboteurs who only break the emergency alarm machines (COX-2, in inflammatory cells) but leave the maintenance machines (COX-1, in stomach/platelets) alone.
Analogy 2: Aspirin and the Suicide Mission
Most NSAIDs are like someone sitting on the factory machine to stop it. They get up, the machine starts again. Reversible.
Aspirin is like a saboteur who pours superglue into the machine and then leaves. The machine is destroyed forever. Irreversible. A new machine has to be built (takes time).
For platelets (which cannot build new machines because they have no factory - no nucleus), aspirin's damage is permanent for the platelet's entire lifetime (7-10 days).
Analogy 3: COX-1 vs COX-2 - The Two Fire Stations
COX-1 = The old, permanent fire station that runs all the time. It handles routine calls: protecting the stomach, helping platelets form blood clots, maintaining kidney blood flow. Essential everyday.
COX-2 = A temporary emergency fire station opened only during a crisis (inflammation, injury). It handles the inflammation response.
Non-selective NSAIDs = Shut down BOTH fire stations. Inflammation is controlled, but now the stomach has no protection and platelets cannot clump.
Coxibs = Shut down only the emergency station (COX-2). Inflammation controlled; the everyday station (COX-1) still runs - stomach protected, platelets still work.
BUT - the emergency station (COX-2) also made a fire-retardant chemical (PGI2) that prevented the dry wood (platelets + blood vessels) from catching fire (clotting). Shut it down, and the wood can catch fire (thrombosis risk).
Analogy 4: The Reye's Syndrome Story (Aspirin in Children)
Imagine a child's liver is a power plant. When a virus infects the child, the power plant is already working overtime trying to fight the infection. Now you give aspirin - it's like throwing a spanner into the power plant's engine while it's already strained. The engine (mitochondria) breaks down. The power plant (liver) catches fire. The brain loses power (encephalopathy). This is Reye's syndrome.
Analogy 5: Aspirin vs Ibuprofen and Antiplatelet Protection
Imagine COX-1 in a platelet is a parking spot reserved for aspirin (aspirin's antiplatelet effect). Aspirin parks there permanently (irreversible).
Ibuprofen is a bigger car that parks in the same spot. If ibuprofen parks there first (patient takes ibuprofen before aspirin), it blocks aspirin from getting in. When ibuprofen eventually drives away (reversible), the spot is empty again - aspirin missed its chance. The platelet's COX-1 is now unmodified, and TxA2 is made freely.
This is why patients on low-dose aspirin for heart protection should NOT take ibuprofen simultaneously.
Analogy 6: The Kidney Prostaglandin Story
The kidney's blood vessels are like a garden hose with a squeeze. Normally, without NSAIDs, your body makes prostaglandins that OPEN (relax) the hose - water (blood) flows freely.
Now imagine you are dehydrated or in heart failure. Your body is already in crisis mode - squeezing all the hoses to maintain blood pressure. The prostaglandins are now working VERY HARD to keep the kidney hose open.
Give an NSAID → block prostaglandins → kidney hose suddenly gets squeezed shut → no blood to kidney → AKI.
In a healthy, well-hydrated person, the NSAID-induced loss of prostaglandins is not as dangerous. But in heart failure, dehydration, or combined with ACE inhibitors/diuretics - it is the "triple whammy" that causes catastrophic AKI.
SECTION 5: STEP-BY-STEP CLINICAL REASONING
Scenario 1: A 25-year-old woman with severe menstrual cramps
How does a doctor think?
Step 1: What is causing the pain?
During menstruation, endometrial cells shed and release large amounts of prostaglandins (especially PGF2α and PGE2). These cause uterine muscle to contract strongly and also sensitize pain fibers → cramping pain.
Step 2: What drug should address the mechanism?
We need to reduce prostaglandins. COX inhibitors (NSAIDs) are ideal.
Step 3: Which NSAID?
- Ibuprofen: Effective, well-tolerated, available OTC - first choice
- Mefenamic acid: Good choice because it both inhibits COX AND blocks prostaglandin receptors - dual mechanism
- Naproxen: Longer-acting, so fewer doses needed
- Avoid: Indomethacin (too toxic for this use)
Step 4: When to start?
Start 1-2 days before expected onset of menses (to prevent prostaglandin surge) - not just when pain starts.
Step 5: Safety considerations?
- Is she pregnant? NSAIDs can cause premature ductus closure in third trimester
- Does she have asthma? AERD risk
- Does she have a history of GI ulcer? Use with PPI or consider paracetamol
Scenario 2: A 65-year-old man with acute gout, knee pain
Step 1: What is going on?
Uric acid crystals deposited in knee joint → phagocytes engulf crystals → release IL-1β, TNF, prostaglandins → intense inflammation, swelling, redness, excruciating pain
Step 2: What do we need?
Rapid, potent anti-inflammatory action.
Step 3: NSAID choice?
- Indomethacin 25-50 mg three times daily - historically preferred for acute gout (most potent, fastest onset)
- Ibuprofen high dose: Also effective
- Naproxen: Acceptable
Step 4: Patient factors?
- Age 65 → renal function may be reduced → be cautious with NSAIDs (AKI risk)
- Heart disease? → cardiovascular risk of NSAIDs
- Peptic ulcer history? → add PPI or use colchicine/steroids instead
- On warfarin? → avoid NSAIDs (bleeding risk)
Step 5: Alternatives if NSAIDs contraindicated?
- Colchicine (blocks neutrophil migration)
- Oral prednisolone
- IL-1 inhibitors (anakinra, canakinumab) for refractory cases
Scenario 3: A 55-year-old with knee osteoarthritis and a history of peptic ulcer
The dilemma: Needs NSAIDs for joint pain, but has a history of peptic ulcer.
Step 1: Can we use a non-drug approach first? Weight loss, physiotherapy, topical diclofenac gel (minimal systemic absorption).
Step 2: If oral NSAID needed: Use celecoxib (COX-2 selective - spares gastric COX-1) + add omeprazole (PPI) for maximum gastric protection.
Step 3: Test and treat H. pylori if not done (H. pylori + NSAID = highest ulcer risk).
Step 4: Use lowest effective dose for shortest duration. Regular monitoring of renal function.
Scenario 4: Patient who took "many aspirin" for pain - suspected overdose
Think through:
- Respiratory alkalosis first (direct stimulation of respiratory center → hyperventilation)
- Then metabolic acidosis (accumulation of lactic acid, salicylic acid)
- Combined = mixed respiratory alkalosis + metabolic acidosis (classic high-yield exam finding)
Management:
- Urine alkalinization (IV sodium bicarbonate) → increases urinary pH → ionizes salicylate → trapped in urine → increased excretion
- Activated charcoal if within 2 hours of ingestion
- Dialysis for severe toxicity (salicylate level >700 mg/L, or severe metabolic acidosis, or AKI)
- Do NOT acidify plasma (worsens toxicity - more drug enters brain)
SECTION 6: MEMORY TOOLS
Mnemonic 1: NSAID Adverse Effects - "GI CARB"
G - Gastric ulceration / GI bleeding
I - Interstitial nephritis / increased renal toxicity
C - Cardiovascular risk (MI, stroke, hypertension)
A - Aspirin: Reye's syndrome in children, antiplatelet effect
R - Renal effects (AKI, water retention, hyperkalemia)
B - Bronchospasm (AERD - aspirin-exacerbated respiratory disease)
Mnemonic 2: Contraindications - "ARCH-3 PEEL"
A - Active peptic ulcer
R - Renal failure
C - Children + viral illness (aspirin → Reye's)
H - Hepatic failure (severe)
3 - Third trimester of pregnancy
P - Platelet disorders / anticoagulant use
E - Elderly (use with caution)
E - AERD / asthma
L - Low GFR states (heart failure, dehydration)
Mnemonic 3: Aspirin Toxicity - "THERMAL TRIP"
Early salicylism: Tinnitus, Hyperthermia (late), Epiphorea (tears/GI irritation), Respiratory alkalosis (FIRST acid-base change), Metabolic acidosis (LATER), Agitation/confusion, Liver dysfunction (Reye's in kids)
Mnemonic 4: COX-1 Functions - "HELP"
H - Hemostasis (TxA2 in platelets)
E - Epithelium protection (gastric mucosa)
L - Low-flow maintenance (renal vasodilation)
P - Platelet aggregation
These are what you LOSE when you block COX-1 non-selectively.
Memory Table: Drug Half-Lives (Clinically Important)
| Drug | Half-life | Dosing Frequency |
|---|
| Aspirin | 15-20 min (aspirin itself); salicylate 2-30 hrs depending on dose | Multiple times daily |
| Ibuprofen | ~2 hours | 3-4 times daily |
| Diclofenac | 1-2 hours | 2-3 times daily |
| Ketorolac | ~5-6 hours | 4-6 hourly (max 5 days) |
| Naproxen | ~14 hours | Twice daily |
| Celecoxib | ~11 hours | Once or twice daily |
| Meloxicam | ~20 hours | Once daily |
| Piroxicam | ~45-50 hours | Once daily |
Visual Memory: Aspirin's Unique Irreversible Effect
Regular NSAID:
Drug molecule -----> COX enzyme (reversible block)
Remove drug ---------> COX enzyme works again
Effect lasts: as long as drug is in blood
Aspirin:
Aspirin -----> Acetylates COX enzyme (COVALENT, IRREVERSIBLE)
Remove aspirin --> COX enzyme STILL DEAD
|
New COX enzyme must be synthesized
Nucleated cells: recover in hours
Platelets (no nucleus): NEVER recover
Platelet effect lasts 7-10 days (platelet lifespan)
Comparison Table: Non-selective vs COX-2 Selective NSAIDs
| Feature | Non-selective NSAIDs | COX-2 Selective (Coxibs) |
|---|
| GI ulcer risk | HIGH | Low |
| Platelet effect | Inhibit aggregation | Minimal/None |
| CV/thrombosis risk | Moderate-high (especially diclofenac, ibuprofen) | Higher (due to PGI2 loss) |
| Renal effects | Present | Present (COX-2 in kidney) |
| Can use with low-dose aspirin? | With caution (ibuprofen competes) | Yes (celecoxib spares COX-1) |
| Asthma/AERD risk | Yes | Yes (some risk) |
| Cost | Low (generic) | Higher |
| Examples | Ibuprofen, naproxen, indomethacin | Celecoxib, etoricoxib |
SECTION 7: EXAMINER'S CORNER
Most Tested Facts
- Mechanism of action: COX inhibition → reduced prostaglandin synthesis
- Aspirin is the ONLY irreversible COX inhibitor (acetylation)
- Aspirin is contraindicated in children with viral illness (Reye's syndrome)
- Coxibs spare COX-1 → less GI toxicity but NO protection from renal toxicity or CV risk
- Celecoxib has INCREASED cardiovascular thrombotic risk (PGI2 inhibition without TxA2 inhibition)
- Aspirin toxicity: mixed respiratory alkalosis + metabolic acidosis
- Aspirin blocks platelet TxA2 permanently for 7-10 days
- Ibuprofen can antagonize aspirin's antiplatelet effect (competition for COX-1 binding)
- NSAIDs contraindicated in third trimester (premature ductus closure)
- AERD/Samter's Triad: Asthma + nasal polyps + aspirin sensitivity
- Indomethacin is used to close patent ductus arteriosus in neonates
- Ketorolac is the only NSAID available parenterally (for short-term use)
- Paracetamol hepatotoxicity → NAPQI → treat with N-acetylcysteine
Most Likely Essay Questions
- "Write a note on NSAIDs: mechanism of action, adverse effects, and contraindications."
- "Discuss the pharmacological basis of using aspirin as an antiplatelet agent. Why is it contraindicated in children?"
- "Compare and contrast non-selective NSAIDs and COX-2 selective inhibitors."
- "Discuss the adverse effects of NSAIDs, explaining the underlying mechanism for each."
- "Write about the management of aspirin poisoning."
- "Explain the concept of aspirin-exacerbated respiratory disease (AERD)."
Most Likely Short Notes
- Aspirin (mechanism + unique properties + uses + toxicity)
- Celecoxib / COX-2 selective inhibitors
- Indomethacin
- Paracetamol / acetaminophen toxicity
- Prostaglandins and their pharmacological significance
- Reye's syndrome
Most Likely Viva Questions
- "What is the mechanism of action of aspirin as an analgesic vs antiplatelet?"
- "Why does aspirin cause Reye's syndrome in children?"
- "Why do COX-2 selective inhibitors cause cardiovascular side effects?"
- "What is the acid-base disturbance in aspirin poisoning?"
- "Why can't aspirin's antiplatelet effect be reversed easily?"
- "How does ibuprofen interact with aspirin?"
- "Why are NSAIDs contraindicated in the third trimester of pregnancy?"
- "What is AERD? What is the mechanism?"
Most Likely MCQs
-
A patient is on low-dose aspirin for MI prevention. He starts taking ibuprofen for back pain. The likely consequence is:
Answer: Reduced antiplatelet effect of aspirin (ibuprofen competitively blocks COX-1, preventing aspirin's irreversible acetylation)
-
A child with chickenpox is given aspirin for fever. The most dangerous complication is:
Answer: Reye's syndrome (encephalopathy + hepatic failure)
-
The mechanism of aspirin-exacerbated respiratory disease is:
Answer: Shunting of arachidonic acid to the lipoxygenase pathway → increased leukotrienes → bronchoconstriction
-
Which NSAID is used to close patent ductus arteriosus?
Answer: Indomethacin (or ibuprofen in some protocols)
-
Aspirin in toxic doses causes which acid-base disturbance?
Answer: Mixed respiratory alkalosis + metabolic acidosis
-
The antidote for paracetamol toxicity is:
Answer: N-acetylcysteine (NAC)
-
COX-2 selective inhibitors cause increased cardiovascular risk because:
Answer: They inhibit PGI2 (prostacyclin) production without inhibiting platelet TxA2 → pro-thrombotic imbalance
-
A 70-year-old man with heart failure is given an NSAID for joint pain. What renal complication is most likely?
Answer: Acute kidney injury (AKI) - prostaglandins maintaining renal blood flow are blocked
Common Traps Students Fall Into
TRAP 1: "Aspirin blocks COX-2 in inflammation - so it should be the safest NSAID." WRONG. Aspirin irreversibly blocks COX-1 (especially in platelets and stomach) and COX-2 both. Its GI and platelet effects are significant.
TRAP 2: "COX-2 inhibitors are totally safe for the GI tract." NOT TOTALLY. They reduce peptic ulcer risk significantly (clinical trials show ~50% reduction) but do not eliminate it entirely, especially with high doses. They still carry renal risks.
TRAP 3: "Paracetamol has no side effects since it doesn't inhibit COX peripherally." WRONG. Paracetamol has a narrow therapeutic index. In overdose (even relatively modest overdose in alcoholics or malnourished patients), it causes fatal hepatic necrosis.
TRAP 4: "Ibuprofen is completely safe in children." Ibuprofen IS safe (and preferred over aspirin) for fever in children, but should be used with caution in children under 6 months, and should be dose-adjusted by weight.
TRAP 5: "Aspirin toxicity = metabolic acidosis only." WRONG. The FIRST change is respiratory ALKALOSIS (direct respiratory center stimulation). Later, metabolic acidosis supervenes. The full picture = mixed disorder.
TRAP 6: "NSAIDs don't affect the kidney much." NSAIDs are a major cause of AKI, especially in at-risk populations. COX-2 selective inhibitors also cause renal effects because COX-2 is expressed in the kidney.
TRAP 7: "Celecoxib can be given freely to patients with sulfa allergy." WRONG. Celecoxib has a sulfonamide moiety. Avoid in sulfonamide allergy.
SECTION 9: HIGH-YIELD REVISION SHEET
ONE-PAGE RAPID REVIEW: NSAIDs
DEFINITION: Drugs that reduce pain, fever, and inflammation by inhibiting cyclooxygenase (COX) enzymes → reducing prostaglandin synthesis.
MECHANISM:
- COX enzyme: converts arachidonic acid → PGG2 → PGH2 → prostaglandins, TxA2, PGI2
- NSAIDs block COX → reduce all downstream prostanoids
- COX-1 = constitutive (housekeeper: stomach protection, platelet TxA2, renal blood flow)
- COX-2 = inducible (inflammation, fever, COX-2 in endothelium makes PGI2)
ASPIRIN IS UNIQUE:
- Irreversible COX inhibitor (acetylation)
- Antiplatelet effect: permanent for platelet lifetime (7-10 days)
- Dose-dependent: low → antiplatelet; medium → analgesic/antipyretic; high → anti-inflammatory
- Contraindicated in children with viral illness (Reye's syndrome)
- Toxicity: mixed respiratory alkalosis + metabolic acidosis; tinnitus
COXIBS (celecoxib, etoricoxib):
- Selective COX-2 inhibitors
- Less GI toxicity (spare gastric COX-1)
- No platelet inhibition (spare platelet COX-1)
- INCREASED cardiovascular risk (block PGI2 but not TxA2)
- Celecoxib: contraindicated in sulfonamide allergy
- Renal effects STILL occur (COX-2 expressed in kidney)
INDOMETHACIN:
- Most potent anti-inflammatory NSAID
- Most GI toxic
- Closes PDA in neonates
- Drug of choice for: acute gout, pericarditis (with aspirin/colchicine), ankylosing spondylitis
KETOROLAC:
- Only widely used parenteral NSAID
- Maximum 5 days use
- Potent analgesic
MEFENAMIC ACID:
- Preferred for dysmenorrhea
- Blocks COX AND prostaglandin receptors
PARACETAMOL:
- NOT a classic NSAID (no peripheral anti-inflammatory effect)
- Centrally acting antipyretic/analgesic
- Toxicity: NAPQI → hepatic centrilobular necrosis
- Antidote: N-acetylcysteine (NAC)
KEY ADVERSE EFFECTS:
- GI: peptic ulcer, GI bleeding (COX-1 inhibition in stomach)
- Cardiovascular: MI, stroke, hypertension (especially coxibs)
- Renal: AKI, water/Na+ retention, hyperkalemia, interstitial nephritis
- Platelet: bleeding tendency (non-selective; not coxibs)
- AERD/Aspirin-sensitive asthma: leukotriene surge
- Reye's syndrome (aspirin in viral illness in children)
KEY CONTRAINDICATIONS:
- Active peptic ulcer, severe renal/hepatic failure
- Third trimester pregnancy (premature PDA closure)
- Children with viral illness (aspirin only - Reye's)
- Asthma (caution - AERD)
- Celecoxib: sulfonamide allergy
KEY INTERACTIONS:
- Warfarin: increased bleeding
- ACE inhibitors + diuretics + NSAIDs = "triple whammy" AKI
- Lithium: toxicity
- Methotrexate: toxicity
- Ibuprofen antagonizes low-dose aspirin
PROSTAGLANDINS REVIEW:
- PGE2: pain sensitization, fever, gastric protection, vasodilation
- PGI2 (prostacyclin): anti-platelet, vasodilation, renal maintenance
- TxA2: pro-platelet, vasoconstriction (from platelets)
- PGF2α: uterine contraction (important for dysmenorrhea)
SECTION 10: SELF-ASSESSMENT
10 Short-Answer Questions with Explanations
Q1. Explain why aspirin's antiplatelet effect lasts 7-10 days but ibuprofen's antiplatelet effect only lasts a few hours.
Answer:
Aspirin IRREVERSIBLY inhibits COX-1 by covalently acetylating it. Platelets are anucleate (no nucleus), so they cannot synthesize new COX-1 protein. The COX-1 remains permanently inactivated for the entire lifespan of the platelet (7-10 days).
Ibuprofen reversibly inhibits COX-1 (competitive block). Once ibuprofen is metabolized and cleared from the platelet (a few hours), COX-1 resumes function. The antiplatelet effect therefore mirrors the drug's plasma half-life (~2 hours).
Q2. A 45-year-old asthmatic patient is given aspirin for a headache and develops acute severe bronchospasm. What is the mechanism?
Answer:
This is Aspirin-Exacerbated Respiratory Disease (AERD). Aspirin blocks COX enzymes, preventing arachidonic acid from entering the prostaglandin pathway. Arachidonic acid is then redirected ("shunted") into the lipoxygenase pathway, producing large amounts of cysteinyl leukotrienes (LTC4, LTD4, LTE4). Leukotrienes are powerful bronchoconstrictors and cause the acute asthma attack. Patients with asthma AND nasal polyps are particularly at risk. All non-selective NSAIDs can trigger this, as can coxibs to a lesser extent.
Q3. Why is aspirin contraindicated in children with chickenpox or influenza?
Answer:
Giving aspirin to children under 12 years with viral infections (especially chickenpox/influenza) can cause Reye's syndrome - a rare but potentially fatal illness characterized by acute non-inflammatory encephalopathy and fatty liver degeneration. The exact mechanism is not fully defined, but aspirin appears to impair mitochondrial function in hepatocytes already stressed by viral infection, leading to hepatic failure and secondary brain involvement. Paracetamol or ibuprofen should be used instead for fever in children.
Q4. Explain the acid-base disturbance seen in aspirin toxicity.
Answer:
There are two sequential phases:
- Early/Mild toxicity: Aspirin directly stimulates the medullary respiratory center → hyperventilation → increased CO2 elimination → respiratory alkalosis (low pCO2, high pH). This is the FIRST acid-base disturbance.
- Late/Severe toxicity: Aspirin uncouples oxidative phosphorylation in mitochondria → cellular metabolism shifts to anaerobic → lactic acid accumulates. Additionally, salicylic acid itself is an organic acid. These together produce metabolic acidosis (low pH, low bicarbonate, increased anion gap).
The final picture in severe poisoning is: mixed respiratory alkalosis + metabolic acidosis = classic exam finding.
Q5. Why do COX-2 selective inhibitors (coxibs) increase the risk of myocardial infarction?
Answer:
In normal vasculature, there is a balance between:
- TxA2 (made by platelets via COX-1): promotes platelet aggregation and vasoconstriction
- PGI2/Prostacyclin (made by vascular endothelium via COX-2): inhibits platelet aggregation and causes vasodilation
COX-2 selective inhibitors (like celecoxib) block endothelial COX-2 → reduce PGI2 production. However, they do NOT block platelet COX-1, so TxA2 production continues unchecked. The balance tips toward TxA2 dominance → a pro-thrombotic, vasoconstrictive state → increased risk of coronary thrombosis → myocardial infarction. This is why rofecoxib was withdrawn from the market after the VIGOR trial showed doubling of MI risk.
Q6. How does indomethacin close patent ductus arteriosus in premature neonates?
Answer:
The ductus arteriosus is a fetal blood vessel that bypasses the lungs (connecting pulmonary artery to aorta). It normally closes after birth. In premature neonates, it may remain open (patent). Prostaglandin E2 (PGE2) actively keeps the ductus arteriosus open (vasodilatory effect on ductal smooth muscle). By inhibiting COX → reducing PGE2 → ductal smooth muscle constricts and the ductus closes. Indomethacin is the traditional drug; ibuprofen IV is now also used. This is one of the most clinically important uses of NSAIDs in neonatology.
Q7. A patient on low-dose aspirin for heart protection also takes ibuprofen for arthritis. Why is this a problem?
Answer:
Both aspirin and ibuprofen compete for the same binding site on COX-1 (the hydrophobic substrate-binding channel). If the patient takes ibuprofen first, ibuprofen molecules occupy and reversibly block the COX-1 active site. Aspirin, which needs to physically access and acetylate the active site to achieve its irreversible antiplatelet effect, cannot gain access while ibuprofen is bound. When ibuprofen is eventually cleared, COX-1 is unmodified (ibuprofen's block was reversible), and TxA2 production resumes. Aspirin's irreversible antiplatelet protection is therefore never achieved. The solution: take aspirin at least 30-60 minutes BEFORE ibuprofen (to allow aspirin to acetylate first), or use a different analgesic like paracetamol or a coxib that does not compete with COX-1.
Q8. Why are NSAIDs contraindicated in the third trimester of pregnancy?
Answer:
Three main reasons:
- Premature closure of the ductus arteriosus: In the fetus, PGE2 keeps the ductus arteriosus open (needed for fetal circulation to bypass lungs, which are not yet used). NSAID inhibition of prostaglandins can cause premature ductal closure → fetal cardiac failure and pulmonary hypertension.
- Oligohydramnios: NSAIDs reduce fetal urine production (prostaglandins maintain renal blood flow in the fetus) → reduced amniotic fluid → fetal complications.
- Delayed/prolonged labor: Prostaglandins (especially PGF2α) are essential for initiating and maintaining uterine contractions during labor. NSAIDs reduce them → tocolytic effect → prolonged labor.
Q9. What is the mechanism of paracetamol hepatotoxicity, and how does N-acetylcysteine (NAC) treat it?
Answer:
Normal metabolism: Paracetamol is metabolized in the liver mostly by conjugation with glucuronide and sulfate (safe pathways), and a small amount by CYP2E1 to a toxic intermediate, NAPQI (N-acetyl-p-benzoquinoneimine). NAPQI is rapidly detoxified by conjugation with glutathione.
In overdose: Glucuronide/sulfate pathways are saturated. More paracetamol is forced through CYP2E1. NAPQI production overwhelms glutathione stores. Free NAPQI covalently binds to liver cell proteins → causes oxidative damage and cell death → centrilobular hepatic necrosis (zone 3, where CYP2E1 is concentrated).
NAC mechanism: NAC replenishes hepatic glutathione (it is a glutathione precursor - provides cysteine for glutathione synthesis) AND can directly react with NAPQI. This restores the capacity to detoxify NAPQI. NAC is most effective when given within 8-10 hours of ingestion, but can still benefit up to 24-36 hours later.
Q10. A 70-year-old man with chronic heart failure and osteoarthritis is prescribed an NSAID. List all the reasons why this combination is dangerous.
Answer:
Multiple simultaneous risks:
-
AKI: Heart failure → reduced cardiac output → kidneys rely heavily on prostaglandins (PGE2, PGI2) to maintain renal blood flow by dilating afferent arterioles. NSAIDs block these prostaglandins → afferent arteriole constriction → GFR falls → acute kidney injury.
-
Worsening heart failure: NSAIDs cause sodium and water retention (prostaglandins normally help the kidney excrete Na+). NSAID use → Na+ and water retention → increased plasma volume → increased preload and afterload → worsening heart failure.
-
Hypertension: Same Na+/water retention mechanism can elevate blood pressure, further straining the failing heart.
-
Interaction with heart failure medications: Most CHF patients are on ACE inhibitors/ARBs (which reduce angiotensin II, itself a compensatory vasoconstrictor). ACE inhibitors + NSAIDs = "double blockade" of renal autoregulation → AKI. If also on diuretics: NSAIDs reduce the effect of diuretics AND add to renal toxicity = "triple whammy" AKI.
-
Cardiovascular thrombotic risk: NSAIDs (especially coxibs and diclofenac) increase MI and stroke risk - dangerous in a compromised heart.
Alternative approach: Use paracetamol for pain (does not affect prostaglandins peripherally, no CV or renal risks at therapeutic doses). Consider topical NSAIDs for local joint pain. If oral NSAIDs absolutely necessary: lowest dose, shortest duration, monitor renal function and electrolytes closely.
This complete learning note has been compiled from authoritative sources including Goodman & Gilman's The Pharmacological Basis of Therapeutics, Katzung's Basic and Clinical Pharmacology (16th Edition), Lippincott Illustrated Reviews: Pharmacology, Robbins and Cotran Pathologic Basis of Disease, Brenner and Rector's The Kidney, and Miller's Anesthesia.
End of NSAIDs Complete Learning Note