Pulmonary Hypertension
Reading File
Finding Sources
Searching PubMed
"pulmonary hypertension"[MeSH Terms] AND ("diagnosis" OR "management")
Searching Images
pulmonary hypertension pathophysiology diagram
Reading File
Finding Sources
Searching PubMed
"pulmonary arterial hypertension"[MeSH Terms] AND "treatment"[MeSH Terms]
Reading File
Finding Sources
Searching PubMed
"pulmonary hypertension" AND "guidelines"
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Pulmonary Hypertension
Definition
Pulmonary hypertension (PH) is defined by a pathologic elevation of mean pulmonary artery pressure (mPAP) > 20 mmHg at rest, measured by right heart catheterization (RHC). The threshold was revised downward from the traditional ≥25 mmHg. The term pulmonary arterial hypertension (PAH) specifically denotes PH caused by elevated pulmonary vascular resistance (PVR) not secondary to left heart disease or hypoxemic lung disease. PH affects approximately 1% of the global population and consistently carries a poor prognosis regardless of etiology.
- Fuster and Hurst's The Heart, 15th Edition, p. 1742
Classification (WHO/WSPH Groups)
PH is classified into five groups based on major etiology:
| Group | Category | Key Examples |
|---|---|---|
| 1 | Pulmonary arterial hypertension (PAH) | Idiopathic, heritable (BMPR2), CTD-associated, CHD, HIV, porto-PH, drug/toxin-induced |
| 2 | PH due to left heart disease | HFrEF, HFpEF, valvular disease |
| 3 | PH due to lung disease/hypoxia | COPD, ILD, sleep apnea, altitude |
| 4 | Chronic thromboembolic PH (CTEPH) | Post-PE thrombotic occlusion |
| 5 | PH with unclear/multifactorial mechanisms | Sarcoidosis, sickle cell, myeloproliferative disease |
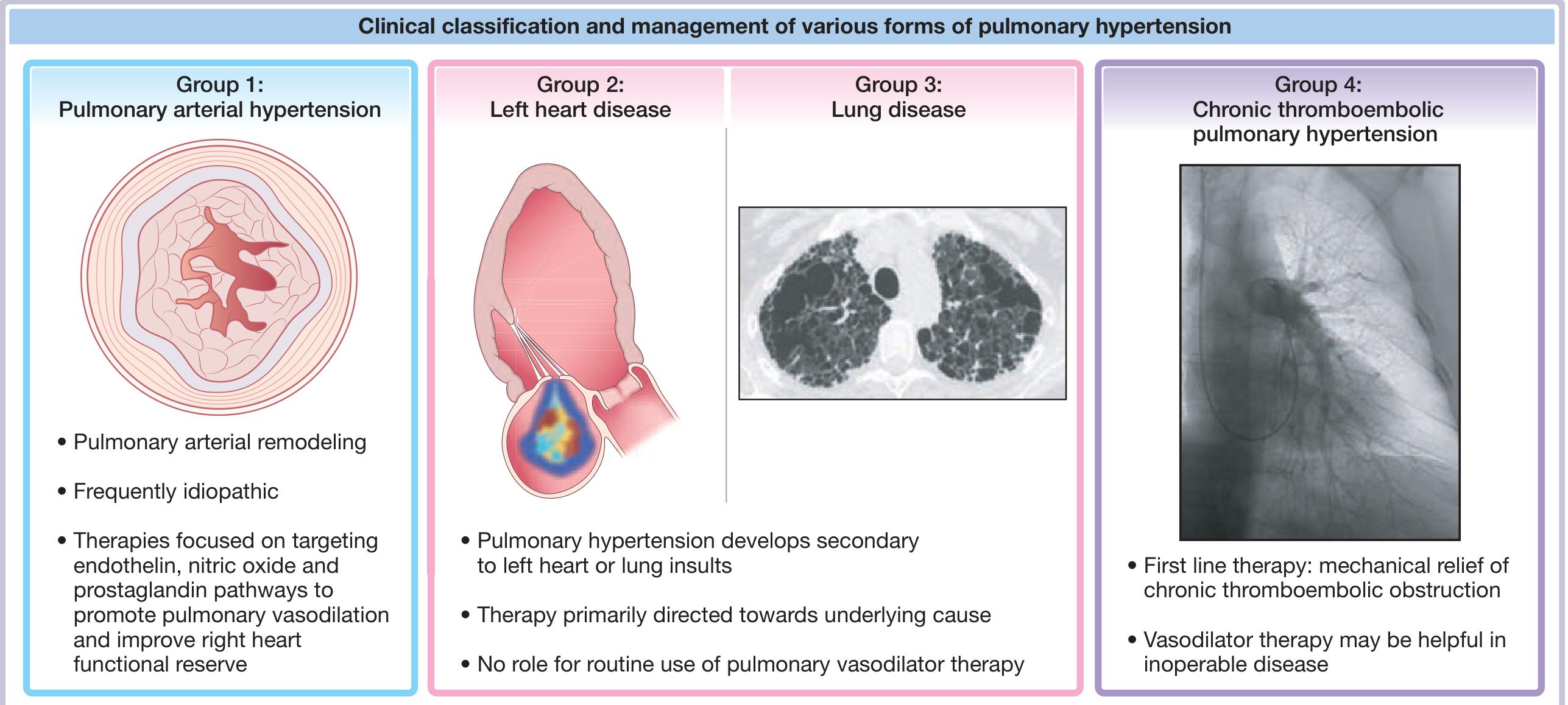
The 6th World Society of Pulmonary Hypertension (WSPH) update introduced notable changes: Group 1.5 (long-term CCB responders), Group 1.6 (PAH with PVOD/PCH features), and split Group 4 into 4.1 (CTEPH) and 4.2 (other pulmonary artery obstructions).
- Fuster and Hurst's The Heart, 15th Edition, Table 57-4
Pathophysiology
The hemodynamic distinction is critical:
- Precapillary PH: elevated mPAP with normal pulmonary artery wedge pressure (PAWP ≤15 mmHg) and elevated PVR — typical of Groups 1, 3, 4
- Postcapillary PH: elevated mPAP with elevated PAWP (>15 mmHg) — typical of Group 2 (left heart disease)
Group 1 PAH Pathobiology
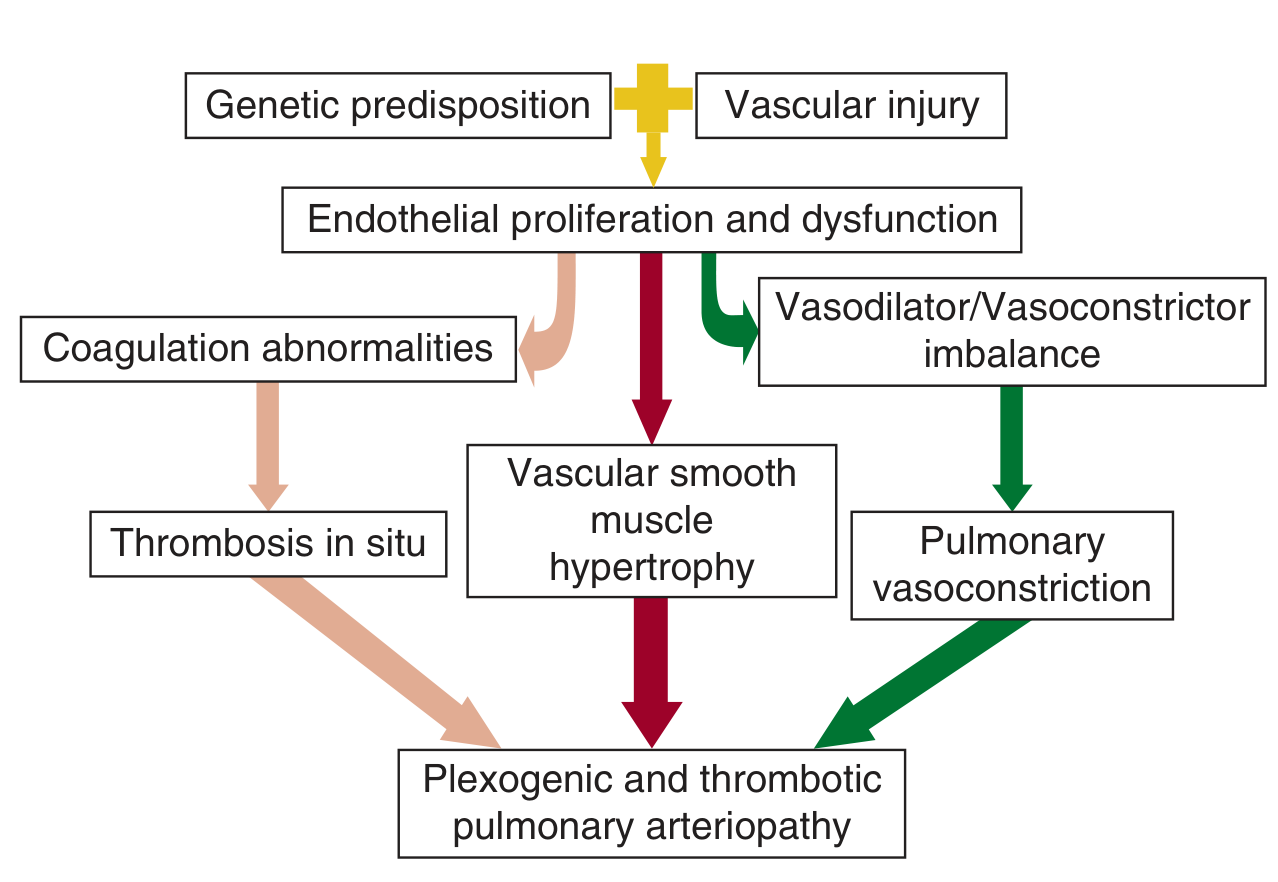
Three converging mechanisms drive progressive PAH:
- Endothelial dysfunction → imbalance of vasodilators (prostacyclin, NO) vs. vasoconstrictors (endothelin-1, thromboxane A2)
- Vascular smooth muscle hypertrophy/proliferation → progressive luminal narrowing
- Thrombosis in situ → coagulation activation and in situ microthrombi
These culminate in plexogenic and thrombotic pulmonary arteriopathy — the hallmark histologic lesion of PAH.
Genetic basis
BMPR2 mutations account for ~15% of idiopathic and ~75% of familial PAH cases. Penetrance is sex-dependent (~14% in males, ~42% in females). Additional rare mutations include ACVRL1, ENG, TBX4, KCNK3, and GDF2. Clonal hematopoiesis of indeterminate potential (CHIP) is an emerging risk factor through systemic inflammation.
- Fuster and Hurst's The Heart, 15th Edition, p. 1749
Group 3 PH (Lung Disease)
Hypoxic pulmonary vasoconstriction, vascular remodeling from polycythemia and increased blood viscosity, and RV pressure overload all contribute. In COPD and IPF, most PH is mild, but a subset develops severe PH disproportionate to the underlying lung disease. PAH-specific therapy is contraindicated in IPF-associated PH — trials showed worsened oxygenation and increased mortality.
Group 4 CTEPH
~4% of acute PE survivors develop CTEPH. Acute thrombus is replaced by chronic intravascular scar. Increased flow shunted through patent vessels raises pressure, and secondary small-vessel remodeling (similar to PAH) further contributes. ~25% of CTEPH cases have no preceding PE history.
Clinical Features
| Symptom/Sign | Mechanism |
|---|---|
| Dyspnea on exertion (earliest) | Reduced CO with exercise |
| Chest pain (substernal, constricting) | RV ischemia; compression of LMCA by dilated PA trunk |
| Syncope/pre-syncope | Fixed low output during exertion |
| Peripheral edema, ascites | Right heart failure |
| Loud P2, RV heave, TR murmur | Pressure-loaded RV |
Chest pain in PH can resemble ischemic angina and may radiate to the neck or arms. In acute massive PE, pain arises from sudden distension of the main PA and mechanoreceptor stimulation.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 889
Diagnosis
Suspected — Non-invasive Workup
| Test | Findings in PH |
|---|---|
| ECG | Right axis deviation, RVH pattern, peaked R in V1, ST depression V1-V3, P pulmonale |
| CXR | Enlarged central PAs, obliteration of retrosternal space, peripheral pruning |
| Echocardiography | RV/RA enlargement, IVS flattening (D-sign), TR jet velocity, RV dysfunction |
| V/Q scan | Screening test for CTEPH (preferred over CTPA for sensitivity) |
| CTPA | Staging anatomic thromboembolic burden in CTEPH |
| 6-minute walk test | Exercise capacity, functional class |
| PFTs + ABG | Identify Group 3 PH |
| Serology | ANA, RF, anti-Scl-70 (connective tissue disease); HIV; LFTs; BNP/NT-proBNP |
| Nocturnal oximetry | Mandatory in all PH patients (even without sleep apnea symptoms) |
Definitive — Right Heart Catheterization (RHC)
RHC is mandatory for confirming PH diagnosis and hemodynamic classification. Required parameters:
- mPAP > 20 mmHg
- PAWP ≤ 15 mmHg (precapillary) vs. > 15 mmHg (postcapillary)
- PVR (calculated)
- Cardiac output
Acute vasodilator testing (with inhaled NO, IV epoprostenol, or IV adenosine) is performed in Group 1 PAH candidates to identify long-term CCB responders (positive response = reduction in mPAP ≥10 mmHg to an absolute value ≤40 mmHg with increased/maintained CO).
- Harrison's Principles of Internal Medicine 22E, pp. 2234–2240; Fuster and Hurst's The Heart, 15th Edition
Treatment
General / Supportive
- Diuretics for RV volume overload
- Supplemental O₂ to keep SpO₂ > 90%
- Supervised exercise/rehabilitation
- Anticoagulation (mandatory in CTEPH; evidence-based in PAH varies by etiology)
- Avoid pregnancy (high mortality risk in PAH)
Group 1 PAH — Targeted Pharmacotherapy
There are 14 FDA-approved therapies across three signaling pathways:
1. Prostacyclin Pathway (↓ prostacyclin → vasoconstriction, thrombosis)
| Drug | Route | Notes |
|---|---|---|
| Epoprostenol | IV (continuous) | Gold standard; improves survival; requires dedicated line |
| Treprostinil | IV, SC, inhaled, oral | More stable; SC associated with site pain |
| Iloprost | Inhaled | 6–9 inhalations/day |
| Selexipag | Oral | Selective IP receptor agonist |
2. Endothelin Receptor Antagonists (↑ ET-1 → vasoconstriction, remodeling)
| Drug | Route | Notes |
|---|---|---|
| Bosentan | Oral | Dual ETA/ETB antagonist; hepatotoxicity monitoring required |
| Ambrisentan | Oral | Selective ETA; fewer liver concerns |
| Macitentan | Oral | Delays clinical worsening (SERAPHIN trial) |
3. NO-cGMP Pathway (↓ NO → vasoconstriction)
| Drug | Route | Notes |
|---|---|---|
| Sildenafil | Oral/IV | PDE5 inhibitor; first-line |
| Tadalafil | Oral | Once-daily PDE5 inhibitor |
| Riociguat | Oral | Soluble guanylyl cyclase stimulator; also approved for CTEPH |
Combination therapy is now standard practice. Early combination (e.g., PDE5i + ERA) is preferred over sequential add-on therapy based on trial data (e.g., AMBITION trial). Riociguat and PDE5 inhibitors must not be combined (risk of severe hypotension).
Among optimally treated PAH patients, estimated survival is ~82% at 1 year, ~67% at 3 years, and ~58% at 5 years.
- Harrison's Principles of Internal Medicine 22E, p. 2240
Group 2 PH (Left Heart Disease)
Therapy directed at the underlying left heart condition. No role for routine pulmonary vasodilator therapy — PAH-specific agents may be harmful by increasing pulmonary venous congestion.
Group 3 PH (Lung Disease)
Treat the underlying lung disease; optimize oxygenation. PAH-specific therapy not routinely indicated (evidence of harm in IPF). Lung transplantation is the treatment of choice for severe PH with end-stage lung disease.
Group 4 CTEPH
-
Lifelong anticoagulation (mandatory)
-
Pulmonary thromboendarterectomy (PTE): standard of care; potentially curative in ~60% of CTEPH patients (proximal disease); 5% in-hospital mortality at experienced centers
-
Balloon pulmonary angioplasty (BPA): for inoperable distal disease; increasing availability
-
Riociguat (oral sGC stimulator): approved specifically for inoperable/residual CTEPH; improves exercise capacity and hemodynamics
-
~20–50% of post-surgical patients retain residual PH from small-vessel disease
-
Fuster and Hurst's The Heart, 15th Edition, pp. 1757–1758
Prognosis & Risk Stratification
Poor prognostic markers include:
- WHO Functional Class III–IV
- 6-MWD < 300–380 m
- Elevated BNP/NT-proBNP
- RV dysfunction on echo (TAPSE, pericardial effusion)
- Hemodynamic deterioration (elevated RAP, low CO, low SvO₂)
- Rapid clinical progression
Key Points Summary
| Issue | Key Fact |
|---|---|
| New diagnostic threshold | mPAP > 20 mmHg (changed from ≥25 mmHg) |
| Diagnostic gold standard | Right heart catheterization |
| CTEPH screening | V/Q scan (not CTPA) |
| PAH first-line | ERA + PDE5i combination |
| CTEPH treatment | PTE surgery (curative); riociguat for inoperable |
| Contraindication | PAH drugs in IPF-associated PH |
| Genetic mutation | BMPR2 (75% familial, 15% idiopathic PAH) |
| Dangerous combination | Riociguat + PDE5 inhibitor (severe hypotension) |
Sources: Fuster and Hurst's The Heart, 15th Edition (Ch. 57); Harrison's Principles of Internal Medicine 22E (Ch. 294); Murray & Nadel's Textbook of Respiratory Medicine; Miller's Anesthesia, 10e; Katzung's Basic and Clinical Pharmacology, 16e
This is a shared conversation. Sign in to Orris to start your own chat.