Cardiomyopathy DCM, HOCM, RCM)
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
dilated hypertrophic restrictive cardiomyopathy comparison diagram
Reading File
Reading File
Reading File
I now have excellent content from Goldman-Cecil and Robbins. Now let me compile the comprehensive answer.
Cardiomyopathies: DCM, HCM (HOCM), and RCM
Overview Comparison
| Feature | DCM | HCM / HOCM | RCM |
|---|---|---|---|
| Wall thickness | Normal or thin | Markedly thickened | Normal or mildly thick |
| Chamber size | Dilated (all 4) | Small/compressed LV cavity | Normal/small ventricles |
| Atria | Dilated (secondary) | Dilated (secondary) | Markedly dilated (biatrial) |
| Systolic function (EF) | Reduced (<40%) | Preserved/hyperdynamic (50–80%) | Normal/near-normal |
| Primary dysfunction | Systolic failure | Diastolic failure | Diastolic failure |
| Sarcomere mutations | Loss-of-function | Gain-of-function | Variable |
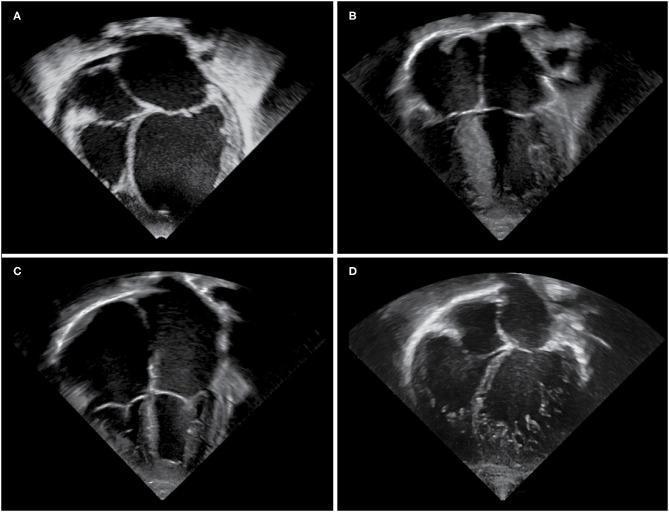
1. Dilated Cardiomyopathy (DCM)
Definition & Epidemiology
DCM is defined by progressive cardiac dilation and contractile (systolic) dysfunction, usually with concurrent hypertrophy, in the absence of coronary artery disease, valvular abnormalities, or pericardial disease. Estimated prevalence is 1 in 250 adults. It is the most common cardiomyopathy in children (up to 58% of pediatric cases). Males and females are approximately equally affected overall.
Pathogenesis & Genetics
- Genetic causes (20–50% of cases): Autosomal dominant is the predominant pattern. Over 50 genes implicated — mostly loss-of-function mutations in cytoskeletal or sarcomere-linking proteins.
- TTN (titin) mutations: most common — ~25% of familial cases, 18% of sporadic cases
- Others: β-myosin heavy chain, α-myosin, cardiac troponin T
- Lamin A/C mutations: cause arrhythmogenic DCM with AV conduction disease preceding heart failure
- X-linked (2–5%): dystrophin gene mutations (Duchenne, Becker, Emery-Dreifuss muscular dystrophies account for 90% of X-linked forms)
- Acquired causes: Viral myocarditis (parvovirus B19, HHV-6, coxsackievirus B, adenovirus), alcohol (>6 drinks/day for 5–10 years; contributes to >10% of US heart failure cases), anthracyclines (overt HF in 5–10% at doses ≥450 mg/m²), peripartum state, hemochromatosis, cocaine, radiation therapy, nutritional deficiencies, sarcoidosis, tachycardiomyopathy
Morphology (Robbins)
- Four-chamber dilation and hypertrophy; heart is flabby and poorly contractile
- Small mural thrombi may form at the LV apex (embolic risk)
- Histology: myocyte hypertrophy + interstitial fibrosis (non-specific); no specific pathologic features at end-stage
Clinical Features
- Most present with symptoms of high pulmonary venous pressure or low cardiac output: exertional dyspnea, orthopnea, PND, fatigue, peripheral edema
- First presentation may be sudden cardiac death or thromboembolic event (AF, LV thrombus → stroke)
- Increasingly diagnosed incidentally on imaging
Diagnosis
- Echo: dilated LV with EF <40%, global hypokinesis
- CMR: dilated chambers, no focal LGE in idiopathic form (vs. ischemic cardiomyopathy which shows subendocardial/transmural scar)
- BNP/NT-proBNP elevated
- Genetic testing for familial cases
Management
- HFrEF guideline-directed therapy: ACE inhibitor/ARB/ARNI + β-blocker + MRA + SGLT2 inhibitor
- ICD for EF <35% after 3 months of GDMT
- CRT (biventricular pacing) if LBBB + QRS ≥150 ms + EF ≤35%
- Anticoagulation for AF or LV thrombus
- Abstinence from alcohol in alcoholic cardiomyopathy (≥50% improve)
- Heart transplantation for refractory cases
Prognosis
5-year survival <50% in severe disease (EF <25%, LVEDD >65 mm, peak VO₂ <12 mL/kg/min).
2. Hypertrophic Cardiomyopathy (HCM) / HOCM
Definition
HCM is characterized by myocardial hypertrophy, defective diastolic filling, and — in one-third of cases — ventricular outflow obstruction (HOCM = hypertrophic obstructive cardiomyopathy). The heart is thick-walled, heavy, and hypercontractile, in striking contrast to DCM.
Pathogenesis & Genetics
- Caused by gain-of-function missense mutations in sarcomeric proteins → myocyte hypercontractility → increased energy use → net negative energy balance
- Autosomal dominant with variable expression; >400 causative mutations identified
- β-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) together with troponin T (TNNT2) account for 70–80% of all HCM cases
- Note: The same genes can cause either HCM (gain-of-function) or DCM (loss-of-function)
Morphology (Robbins)
- Massive myocardial hypertrophy without ventricular dilation
- Asymmetric septal hypertrophy (ASH) in 90% of cases (disproportionate thickening of interventricular septum > LV free wall)
- Concentric hypertrophy in 10%
- LV cavity compressed into a "banana-like" shape on longitudinal section
- Systolic anterior motion (SAM) of the mitral valve: anterior mitral leaflet contacts the septum during systole → LVOT obstruction → subaortic plaque on septal endocardium + mitral leaflet thickening
- Histology: myocyte disarray (hallmark) — haphazardly arranged myocytes with interstitial fibrosis
Outflow Obstruction (HOCM)
- LVOT gradient present at rest or provoked in ~33% of patients
- Worsened by: decreased preload (dehydration, Valsalva, standing), decreased afterload, increased contractility (exercise, digoxin, sympathomimetics)
- Improved by: increased preload (squatting, leg raise), increased afterload (hand grip), decreased contractility (β-blockers, verapamil)
- Classic murmur: harsh crescendo-decrescendo systolic ejection murmur at LLSB, increases with Valsalva and standing, decreases with squatting
Clinical Features
- Wide spectrum: many asymptomatic (incidental echo/ECG finding)
- Symptomatic triad: dyspnea on exertion, angina, syncope
- Sudden cardiac death (SCD): most common cause of SCD in young athletes; arrhythmia-mediated
- Atrial fibrillation common; anti-arrhythmics of choice are disopyramide + β-blocker, or amiodarone
Management
Pharmacological (obstructive form):
- β-blockers (first-line for symptomatic relief)
- Verapamil or diltiazem (if β-blocker not tolerated)
- Disopyramide (added to β-blocker for refractory obstruction)
- Mavacamten (cardiac myosin inhibitor, FDA-approved 2022): reduces LVOT obstruction by decreasing excessive myosin–actin cross-bridge formation
- Avoid: vasodilators, nitrates, diuretics (reduce preload → worsen obstruction), digoxin, sympathomimetics
Invasive (refractory LVOTO):
- Septal myectomy (Morrow procedure): gold standard; surgical resection of proximal septum
- Alcohol septal ablation: catheter-based, injects ethanol into first septal perforator → controlled infarction → reduces septal thickness
SCD prevention:
- ICD implantation for: prior SCA/sustained VT, family history of SCD, massive hypertrophy (wall ≥30 mm), unexplained syncope, NSVT on Holter, abnormal BP response to exercise
3. Restrictive Cardiomyopathy (RCM)
Definition & Epidemiology
RCM is characterized by decreased ventricular compliance (stiffness), impaired diastolic filling, elevated diastolic pressures, and reduced diastolic volume despite normal or near-normal systolic function and wall thickness. It is the least common of the three primary cardiomyopathies.
Causes
| Category | Examples |
|---|---|
| Infiltrative | Amyloidosis (most prevalent), Sarcoidosis |
| Storage disorders | Hemochromatosis, Fabry disease, Glycogen storage diseases |
| Fibrotic | Radiation-induced, Scleroderma, Doxorubicin |
| Endomyocardial | Endomyocardial fibrosis (EMF), Löffler endocarditis (hypereosinophilia) |
| Genetic/Idiopathic | TNNI3, MYH7, DES mutations |
| Metabolic | Carnitine deficiency, fatty acid oxidation defects |
| Miscellaneous | Carcinoid syndrome |
Key Subtypes
Cardiac Amyloidosis (most common secondary RCM in adults):
- Wild-type transthyretin (ATTR) amyloidosis is most prevalent in older patients
- Mutant ATTR: specific V122I mutation in TTR carried by 4% of African Americans → fourfold increased RCM risk
- AL amyloidosis (plasma cell dyscrasia): immunoglobulin light chains directly cardiotoxic
- "Sparkling" myocardium on echo; thick walls with small LV cavity; CMR shows diffuse subendocardial LGE
Endomyocardial Fibrosis (EMF):
- Most common RCM worldwide (tropical Africa, South America, Asia)
- Diffuse fibrosis of ventricular endocardium/subendocardium + tricuspid/mitral valve involvement
- Linked to helminthic infections and nutritional deficiencies
Löffler Endocarditis:
- Associated with peripheral hypereosinophilia + eosinophilic infiltrates
- Eosinophil major basic protein → endocardial/myocardial necrosis → mural thrombus formation → organization/scarring
- No geographic predilection
Morphology (Robbins)
- Ventricles normal size or only slightly enlarged, cavities not dilated
- Myocardium is firm
- Both atria markedly dilated (hallmark) — consequence of restricted ventricular filling
- Microscopy: variable interstitial fibrosis; endomyocardial biopsy often reveals specific etiology
Clinical Features
- Symptoms of heart failure + supraventricular arrhythmias including AF
- Signs of elevated venous pressure: JVP raised, peripheral edema, ascites, hepatomegaly
- Kussmaul's sign (JVP rises on inspiration) — shared with constrictive pericarditis
- Differentiation from constrictive pericarditis is critical (similar hemodynamic profile) but management differs; key tests: CMR (pericardial thickening in CP), endomyocardial biopsy, cardiac catheterization (simultaneous RV/LV pressures)
Diagnosis
- Echo: biatrial dilation with normal ventricular size; restrictive filling pattern (E/A >2, short DT, short IVRT)
- Doppler tissue imaging: annular velocities reduced
- CMR: tissue characterization for amyloid, sarcoid, fibrosis
- Endomyocardial biopsy: often reveals specific etiology
Management
- Treat underlying cause: tafamidis (stabilizes TTR tetramer) for ATTR amyloidosis; phlebotomy for hemochromatosis; steroids for sarcoidosis
- Symptomatic HF: cautious diuresis (small, stiff ventricles are preload-dependent; over-diuresis reduces CO)
- Rate control for AF: prevents tachycardia-mediated filling impairment
- Anticoagulation for AF or intracardiac thrombus
- Transplantation: definitive therapy for refractory primary RCM; contraindicated in systemic amyloidosis (recurs in graft)
Key Differentiating Features at a Glance
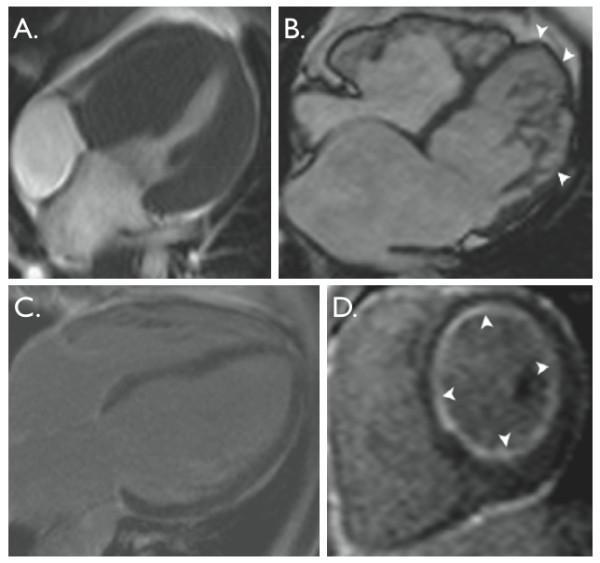
| Feature | DCM | HCM/HOCM | RCM |
|---|---|---|---|
| Mechanism | Systolic dysfunction | Diastolic dysfunction + LVOTO | Diastolic dysfunction (stiff wall) |
| LV size | Dilated | Normal/small | Normal/small |
| Atrial dilation | Mild-moderate | Mild-moderate | Marked (biatrial) |
| Wall thickness | Thin/normal | Markedly increased | Normal |
| EF | Low (<40%) | High (50–80%) | Normal |
| Mural thrombus | Yes (LV apex) | Rare | Yes (eosinophilic) |
| Key mutation | TTN (loss-of-function) | MYH7/MYBPC3 (gain-of-function) | TNNI3/TTR |
| Classic auscultation | S3 gallop, displaced apex | Crescendo-decrescendo systolic murmur ↑ Valsalva | S4, elevated JVP |
| Gold-standard Rx for refractory | Heart transplant | Septal myectomy | Treat etiology; transplant |
| Biopsy finding | Fibrosis, myocyte hypertrophy | Myocyte disarray | Amyloid, fibrosis, eosinophils |
Sources: Robbins & Kumar Basic Pathology 10e, pp. 371–375; Goldman-Cecil Medicine, pp. 514–530; Braunwald's Heart Disease 12e
1,3
Asking for Preferences
Most likely "1, 3" refers to DCM (1) and RCM (3) — a deeper dive into those two. Here they are with full clinical detail:
1. Dilated Cardiomyopathy (DCM)
Definition & Epidemiology
DCM is defined by progressive cardiac dilation + systolic (contractile) dysfunction of the LV or both ventricles, without coronary artery disease, valvular disease, or pericardial disease as the cause.
- Prevalence: 1 in 250 adults
- Most common cardiomyopathy in children (58% of pediatric cases)
- Males = Females overall (except X-linked forms: male predominance)
Pathogenesis
Genetic (20–50% of cases)
| Gene | Protein | Notes |
|---|---|---|
| TTN | Titin | Most common — 25% familial, 18% sporadic |
| LMNA | Lamin A/C | Arrhythmogenic form; AV block precedes HF |
| MYH7 | β-myosin heavy chain | Also mutated in HCM (loss-of-function here) |
| TNNT2 | Troponin T | |
| DMD | Dystrophin | X-linked; Duchenne/Becker muscular dystrophy |
| DES | Desmin | Mixed RCM/DCM + conduction disease |
| FLNC | Filamin C | Lethal ventricular arrhythmias in young adults |
- Autosomal dominant is predominant; X-linked accounts for 2–5%
- All genetic DCM mutations are loss-of-function (contrast with HCM's gain-of-function)
- Because contractile myocytes and conduction fibers share a developmental pathway, conduction abnormalities (LBBB, AV block) are common in inherited DCM
Acquired Causes
| Cause | Mechanism / Notes |
|---|---|
| Viral myocarditis | Parvovirus B19, HHV-6, coxsackievirus B, adenovirus |
| Alcohol | Acetaldehyde directly cardiotoxic; >6 drinks/day × 5–10 yrs; >10% of US HF cases |
| Anthracyclines | Doxorubicin/daunorubicin; overt HF in 5–10% at ≥450 mg/m² |
| Peripartum | Last month of pregnancy to 5 months postpartum |
| Hemochromatosis | Iron overload → myocyte damage |
| Sarcoidosis | Granulomatous infiltration |
| Cocaine | Catecholamine-mediated toxicity + vasospasm |
| Radiation | Diffuse biventricular fibrosis |
| Tachycardiomyopathy | Reversible with rate control |
| Nutritional | Thiamine (beriberi), selenium, carnitine deficiency |
| Autoimmune/inflammatory | SLE, myocarditis |
Morphology
Gross:
- Four-chamber dilation with hypertrophy — heart is flabby, poorly contractile
- Mural thrombi at LV apex (→ systemic emboli/stroke risk)
- Valvular annuli dilated → functional MR/TR
Microscopy (non-specific):
- Myocyte hypertrophy with enlarged nuclei
- Interstitial and replacement fibrosis
- No specific diagnostic histologic pattern at end-stage
Clinical Manifestations
Symptoms (depend on degree of LV dysfunction):
- Exertional dyspnea → orthopnea → PND (rising pulmonary venous pressure)
- Fatigue, exercise intolerance (low cardiac output)
- Palpitations, presyncope, syncope (arrhythmias — AF, VT/VF)
- First presentation may be sudden cardiac death or thromboembolic stroke
- Increasingly found incidentally on imaging (asymptomatic LV dysfunction)
Signs:
- Displaced, diffuse apex beat (cardiomegaly)
- S3 gallop (ventricular filling sound)
- Soft S1, functional MR/TR murmurs (pansystolic)
- Elevated JVP, peripheral oedema, hepatomegaly (right HF)
- Pulmonary crackles (pulmonary oedema)
Investigations
| Test | Finding |
|---|---|
| ECG | LBBB, non-specific ST changes, AF, PVCs |
| CXR | Cardiomegaly, pulmonary venous congestion, Kerley B lines |
| Echo | Dilated LV, EF <40%, global hypokinesis, functional MR, LV thrombus |
| CMR | Dilated chambers; no focal LGE in idiopathic; mid-myocardial LGE in genetic forms (LMNA, FLNC); subendocardial in ischemic |
| BNP/NT-proBNP | Elevated |
| Endomyocardial biopsy | Useful if myocarditis, infiltrative disease, or storage disorder suspected |
| Genetic testing | Recommended in familial/young-onset cases |
| Iron studies/ferritin | Hemochromatosis screen |
| TSH | Thyroid dysfunction |
| Viral serology | Active myocarditis workup |
Management
Guideline-Directed Medical Therapy (GDMT) for HFrEF
- ACEi/ARB → replace with ARNI (sacubitril/valsartan) if tolerated — reduces mortality
- β-blocker (carvedilol, metoprolol succinate, bisoprolol) — reduces mortality
- MRA (spironolactone/eplerenone) — reduces mortality
- SGLT2 inhibitor (dapagliflozin/empagliflozin) — reduces HF hospitalization + CV death
Device Therapy
- ICD: EF ≤35% after ≥3 months optimal GDMT (for SCD prevention)
- CRT (cardiac resynchronization therapy): EF ≤35% + LBBB + QRS ≥150 ms → improves EF, symptoms, mortality
Anticoagulation
- AF → anticoagulate (stroke risk)
- LV thrombus → anticoagulate (embolism risk)
Specific Causes
- Alcoholic: abstinence → ≥50% improve; some normalize EF
- Tachycardiomyopathy: rate/rhythm control → often reversible
- Peripartum: bromocriptine (inhibits prolactin) may help; avoid β-blockers in breast-feeding
- Hemochromatosis: phlebotomy/chelation
- Myocarditis: immunosuppression in giant cell myocarditis
Advanced HF
- LVAD (left ventricular assist device): bridge to transplant or destination therapy
- Heart transplantation: definitive; 1-year survival ~85%, 5-year ~75%
Prognosis
5-year survival <50% in severe disease (EF <25%, LVEDD >65 mm, peak VO₂ <12 mL/kg/min). With modern GDMT, prognosis has significantly improved.
3. Restrictive Cardiomyopathy (RCM)
Definition & Epidemiology
RCM is characterized by ventricular stiffness → impaired diastolic filling → elevated diastolic pressures → reduced diastolic volume despite normal or near-normal systolic function and wall thickness.
- Least common of the primary cardiomyopathies
- Primary (idiopathic) forms are very uncommon; most are secondary to systemic disease
- ~30% of idiopathic RCM is familial (TNNI3, MYH7, DES mutations)
Causes
| Category | Diseases |
|---|---|
| Infiltrative | Amyloidosis (most common), Sarcoidosis |
| Storage disorders | Hemochromatosis, Fabry disease, Glycogen storage diseases |
| Fibrotic | Radiation, Scleroderma, Drugs (doxorubicin, ergotamine, serotonin) |
| Endomyocardial | Endomyocardial fibrosis (EMF), Löffler endocarditis |
| Genetic/sarcomeric | TNNI3 (troponin I), MYH7, DES (desmin) |
| Metabolic | Carnitine deficiency, fatty acid oxidation defects |
| Miscellaneous | Carcinoid syndrome |
Key Subtypes
A. Cardiac Amyloidosis (most prevalent RCM in adults)
- Wild-type ATTR (transthyretin): most common; age-related accumulation; older males predominate
- Mutant ATTR: V122I variant carried by 4% of African Americans → >4× increased risk
- AL amyloidosis: immunoglobulin light chains deposit + directly cardiotoxic; associated with multiple myeloma/MGUS
- Echo: "sparkling" granular myocardium, biatrial dilation, thick walls, small LV cavity, diastolic dysfunction; pericardial effusion
- CMR: diffuse subendocardial LGE — highly specific for amyloid
- Serum free light chains, SPEP, bone marrow biopsy (AL); nuclear scintigraphy with technetium pyrophosphate (ATTR)
B. Endomyocardial Fibrosis (EMF)
- Most common RCM worldwide (tropical: sub-Saharan Africa, India, South America)
- Diffuse fibrosis of ventricular endocardium + subendocardium, often involving tricuspid and mitral valves
- Apical obliteration → reduces chamber volume and compliance
- Linked to helminthic infections (Toxocara?) and nutritional deficiencies
C. Löffler Endocarditis (Hypereosinophilic Syndrome)
- Peripheral hypereosinophilia (>1500 eosinophils/µL for >6 months) + eosinophilic tissue infiltrates
- Mechanism: Eosinophil major basic protein → endocardial/myocardial necrosis → mural thrombus formation → thrombus organization → fibrosis (scarring of endocardium)
- No geographic predilection (contrast with EMF)
- Three phases: necrotic → thrombotic → fibrotic
- Treatment: corticosteroids + hydroxyurea (reduce eosinophilia); anticoagulation; surgical endocardial resection in fibrotic phase
D. Cardiac Sarcoidosis
- Granulomatous infiltration → patchy fibrosis
- Presents with heart block, VT/VF, HF, or SCD
- Steroid therapy; high SCD risk → ICD
Morphology
Gross:
- Ventricles normal size or only slightly enlarged; cavities not dilated
- Myocardium is firm (stiff)
- Both atria markedly dilated — hallmark, due to restricted ventricular filling causing backpressure
- Thrombus in atrial appendages, patchy endocardial fibrosis
Microscopy:
- Variable interstitial fibrosis + myocyte disarray (idiopathic/familial)
- Amyloidosis: extracellular amyloid deposits (Congo red stain → apple-green birefringence under polarized light)
- EMF: dense subendocardial fibrous tissue
- Löffler: eosinophilic infiltrates + mural thrombus
Clinical Manifestations
Symptoms:
- Biventricular heart failure: exertional dyspnea, fatigue, peripheral oedema, ascites
- Supraventricular arrhythmias including AF (common, due to massive atrial dilation)
- Chest pain (if associated with infiltrative disease)
- Syncope/sudden death (sarcoid, amyloid with conduction disease)
- Systemic features of underlying disease (e.g., carpal tunnel syndrome in ATTR amyloid; peripheral neuropathy; macroglossia in AL amyloid)
Signs:
- Raised JVP with Kussmaul's sign (JVP rises on inspiration — failure of RV to accommodate increased venous return)
- S4 gallop (stiff ventricle)
- Hepatomegaly, ascites, peripheral oedema (right HF)
- Pulmonary crackles (left HF)
Investigations
| Test | Finding |
|---|---|
| ECG | Low voltage (amyloid), AV block (sarcoid/ATTR), AF, pseudoinfarct pattern |
| CXR | Normal heart size (small ventricles!) with pulmonary congestion — classic clue |
| Echo | Biatrial dilation, normal/small ventricles, normal EF, restrictive filling (E/A >2, short DT <150 ms, short IVRT), "sparkling" myocardium (amyloid) |
| CMR | Tissue characterization — diffuse LGE (amyloid), patchy LGE (sarcoid), pericardial thickening absent (vs. constrictive pericarditis) |
| Cardiac catheterization | "Square root sign" (dip-and-plateau) on LV pressure tracing; equalization of diastolic pressures; similar to constrictive pericarditis |
| Tc-99m PYP scan | Positive in ATTR amyloidosis (grade 2–3 uptake = diagnostic) |
| Endomyocardial biopsy | Often reveals specific etiology — amyloid (Congo red), eosinophils, granulomas |
| Serum FLC/SPEP/UPEP | AL amyloidosis screen |
RCM vs. Constrictive Pericarditis — Critical Distinction
| Feature | RCM | Constrictive Pericarditis (CP) |
|---|---|---|
| Pericardium (CMR/CT) | Normal | Thickened (>4 mm) |
| Tissue Doppler e' velocity | Reduced (<8 cm/s) | Normal/elevated (>8 cm/s) |
| Respiratory variation on Doppler | Absent or minimal | Present (>25% MV inflow variation) |
| Kussmaul's sign | May be present | Classic |
| LV/RV pressure equalization | Present | Present |
| Endomyocardial biopsy | Abnormal | Normal |
| Treatment | Medical/transplant | Pericardiectomy (curative) |
This distinction is critical because constrictive pericarditis is surgically curable while RCM is not.
Management
| Goal | Intervention |
|---|---|
| Treat etiology | Tafamidis (ATTR amyloid) — stabilizes TTR tetramer, reduces mortality; Phlebotomy (hemochromatosis); Steroids (sarcoid, eosinophilic); Chemotherapy (AL amyloid) |
| Fluid management | Cautious diuresis — stiff ventricles are preload-dependent; over-diuresis → ↓ CO |
| Rate control | Prevent tachycardia → preserve diastolic filling time |
| AF | Anticoagulate; rate/rhythm control |
| SCD prevention | ICD in sarcoid or other high-risk etiologies |
| Advanced disease | Heart transplantation — definitive for primary RCM; contraindicated in systemic AL amyloidosis (disease recurs in graft) |
Prognosis
- Highly etiology-dependent
- Idiopathic RCM: poor prognosis; many progress to end-stage HF requiring transplant
- ATTR amyloid: tafamidis improves survival and quality of life
- AL amyloid: poor without treatment; median survival <6 months untreated
Sources: Robbins & Kumar Basic Pathology 10e, pp. 371–375; Goldman-Cecil Medicine International Edition, pp. 514–530
Can you give definition of cardiomyopathy
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
I have exactly what's needed from Harrison's 22e (2025). Here is a clean, complete answer:
Definition of Cardiomyopathy
Core Definition
Cardiomyopathy describes primary disease of the heart muscle itself, originally excluding myocardial dysfunction resulting from other cardiovascular diseases (e.g., coronary artery disease, valvular disease, hypertensive heart disease, congenital heart disease).
— Harrison's Principles of Internal Medicine, 22e (2025)
In practical clinical usage, however, the term has expanded to also include:
- Ischemic cardiomyopathy (from CAD)
- Valvular cardiomyopathy
- Hypertensive cardiomyopathy
WHO / Functional Definition
Cardiomyopathies are diseases of the myocardium associated with cardiac dysfunction, classified by their functional and structural pattern:
| Type | Core Abnormality | Functional Pattern |
|---|---|---|
| DCM | Dilation + impaired contractility | Systolic dysfunction |
| HCM | Hypertrophy + impaired relaxation | Diastolic dysfunction ± LVOTO |
| RCM | Stiffness + impaired filling | Diastolic dysfunction |
| ARVC | Fibro-fatty replacement (RV > LV) | Arrhythmia + RV dysfunction |
Key Points from the Definition
-
Primary vs. Secondary: True cardiomyopathy is a primary myocardial disorder — not the result of ischemia, valve disease, or hypertension (though secondary forms do exist)
-
Phenotype vs. Etiology: The traditional morphologic classification (DCM, HCM, RCM) is based on structural phenotype, but etiology (genetic, acquired, inflammatory) is increasingly used to guide management
-
Phenotypes overlap: Over time, HCM can evolve to reduced EF ("HCM-rEF") in ~5% of patients; DCM can "reverse remodel" to near-normal EF with treatment
-
Genetic causes are central: Pathogenic variants are being identified that cause heritable cardiomyopathies, sometimes detectable by imaging before clinical disease appears ("preclinical phase")
-
Two-hit model: Clinical expression of a genetic predisposition may be triggered by an additional environmental or acquired insult (e.g., a person with a TTN mutation who also develops viral myocarditis)
WHO 1995 Definition (Classic)
The World Health Organization defined cardiomyopathies as:
"Diseases of the myocardium associated with cardiac dysfunction."
They were divided into:
- Primary (idiopathic): No identifiable systemic or cardiac cause
- Secondary (specific): Myocardial disease as part of a known systemic disorder (amyloidosis, hemochromatosis, sarcoidosis, etc.)
Summary in One Sentence
Cardiomyopathy is a disease of the heart muscle causing structural and/or functional abnormality, in the absence of other cardiovascular conditions sufficient to explain the degree of myocardial dysfunction.
Source: Harrison's Principles of Internal Medicine 22e (2025), Chapter 266 — "Classification of Cardiomyopathy"; Fuster & Hurst's The Heart, 15th Edition
This is a shared conversation. Sign in to Orris to start your own chat.