Describe all types of primary glomerulonephritis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have extensive, detailed content from Harrison's 22E, Robbins Pathologic Basis of Disease, and Comprehensive Clinical Nephrology 7th Ed. I have enough to write a thorough, well-organized answer covering all major primary GN types.
Primary Glomerulonephritis - Complete Overview
Primary (idiopathic) glomerulonephritis refers to glomerular disease where the kidney itself is the primary target, not secondary to a systemic illness. The major types are grouped below.
Overview & Classification
Primary GN presents as either:
- Nephrotic syndrome - massive proteinuria (>3.5 g/day), hypoalbuminemia, edema, hyperlipidemia, lipiduria
- Nephritic syndrome - hematuria (dysmorphic RBCs, RBC casts), hypertension, oliguria, variable proteinuria
- Rapidly progressive (crescentic) pattern - acute loss of renal function over days to weeks
1. Minimal Change Disease (MCD)
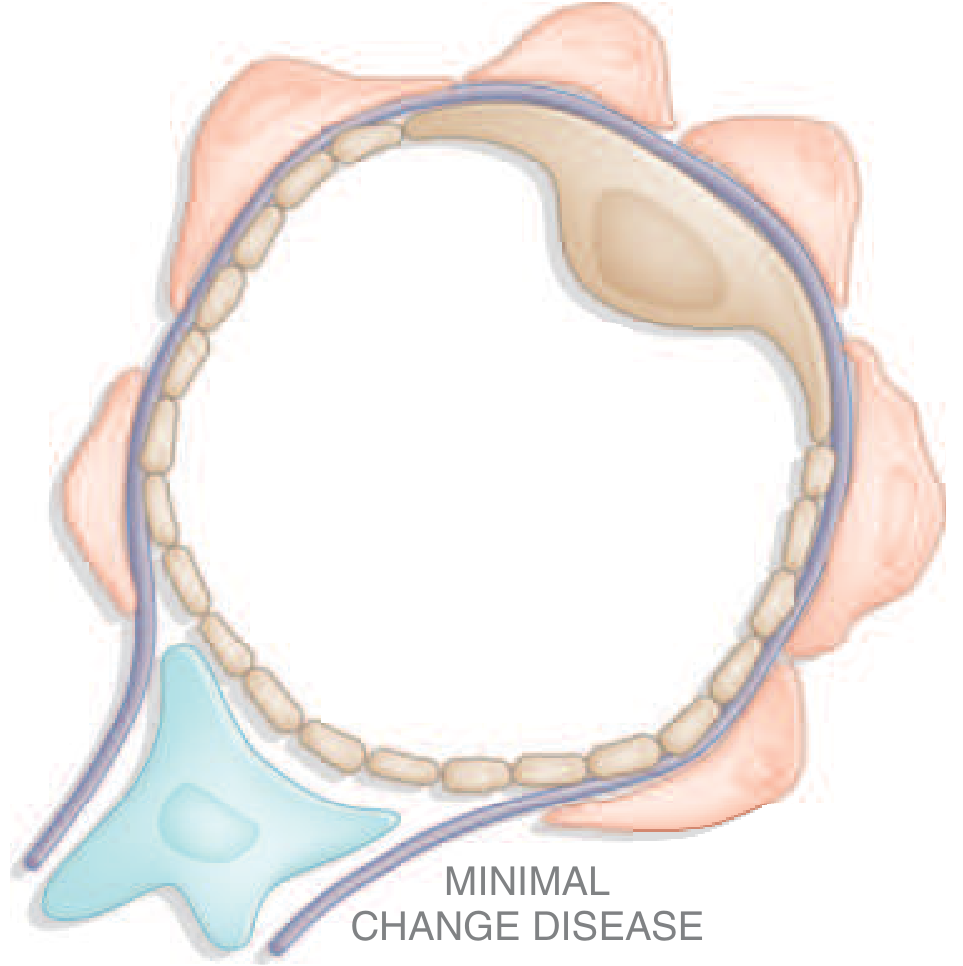
Epidemiology: Most common cause of nephrotic syndrome in children (70-90%); accounts for only 10-15% in adults. Peak incidence at 2-6 years.
Pathogenesis: Formerly attributed solely to T-cell dysfunction causing circulating permeability factors (IL-13, IL-4) and increased CD80 on podocytes. Recent evidence identifies circulating anti-nephrin antibodies (nephrin is a key slit-diaphragm protein), causing podocyte injury and breakdown of the filtration barrier. B-cell involvement is supported by responsiveness to rituximab (anti-CD20).
Morphology:
- Light microscopy: Normal-appearing glomeruli (hence "nil lesion")
- Electron microscopy: Diffuse effacement (flattening/retraction) of podocyte foot processes - the hallmark lesion. No electron-dense deposits; GBM appears normal
- Immunofluorescence: Negative, or subtle mesangial IgM; occasionally fine punctate IgG on podocytes (anti-nephrin antibody binding)
- Proximal tubular cells are loaded with lipid and protein (historical name: lipid nephrosis)
Clinical Features:
- Abrupt onset of nephrotic syndrome; average 24-h urine protein ~10 g; severe hypoalbuminemia
- Acellular urinary sediment (no hematuria, no casts)
- Hypertension in 30% of children, 20-50% of adults
- Selective proteinuria (mainly albumin, especially in children)
- Renal function usually preserved
Treatment & Prognosis:
- Dramatic response to corticosteroids (>90% of children achieve complete remission after 8 weeks); adults respond more slowly but 80-90% achieve remission
- Relapses occur in 70-75% of children after first remission
- Steroid-dependent and frequently relapsing disease responds to rituximab
- Excellent long-term prognosis; even steroid-dependent disease may resolve at puberty
- Harrison's Principles of Internal Medicine 22E; Robbins, Cotran & Kumar Pathologic Basis of Disease
2. Focal Segmental Glomerulosclerosis (FSGS)
Epidemiology: Most common cause of nephrotic syndrome in adults in the United States, particularly in individuals of Hispanic and African descent. Accounts for ~10% of nephrotic syndrome in children and ~35% in adults.
Pathogenesis: Primary FSGS is a diffuse podocytopathy considered to be on the same spectrum as MCD but more severe. Key mechanism involves a circulating permeability factor (possibly suPAR or others not yet fully identified) that injures podocytes diffusely, leading to focal and segmental scarring. High-risk APOL1 alleles (common in those of African ancestry) are a major genetic risk factor, causing more rapid progression to ESKD.
Morphology:
- Sclerosis involves only a segment (not the whole) of only a subset (focal, not all) of glomeruli
- Sclerotic lesions tend to start in the juxtamedullary glomeruli - therefore superficial biopsies may miss the diagnosis
- Podocyte foot process effacement is diffuse (like MCD) even in unaffected glomeruli
- On IF: IgM and C3 in sclerotic segments (non-specific, trapped)
FSGS Variants (Columbia Classification):
| Variant | Key Feature |
|---|---|
| Not Otherwise Specified (NOS) | Most common; generic sclerosis |
| Tip lesion | Sclerosis near tubular pole; best prognosis |
| Perihilar | Sclerosis near vascular pole; often adaptive/secondary |
| Cellular | Endocapillary hypercellularity; aggressive |
| Collapsing | Global capillary collapse; worst prognosis; associated with HIV, APOL1 variants |
Clinical Features:
- Nephrotic syndrome with non-selective proteinuria
- Hypertension, microscopic hematuria, and azotemia are common at presentation
- Unlike MCD: poor response to corticosteroids, progressive course
- At least 50% progress to ESKD over 10 years
- Recurs in ~30% of kidney transplants (primary FSGS much more likely to recur than secondary)
Treatment: Corticosteroids are first-line for primary FSGS; steroid-resistant disease treated with calcineurin inhibitors (cyclosporine, tacrolimus). No role for immunosuppression in secondary, adaptive, or genetic FSGS.
- Harrison's Principles of Internal Medicine 22E; Robbins, Cotran & Kumar Pathologic Basis of Disease
3. Membranous Nephropathy (Membranous GN, MGN)
Epidemiology: Accounts for ~25% of nephrotic syndrome in adults; peak incidence 30-50 years; male:female ratio 2:1. It is the most common cause of nephrotic syndrome in white adults over 40.
Pathogenesis: Chronic immune complex-mediated disease with subepithelial deposits. Major advances:
- PLA2R (M-type phospholipase A2 receptor): Autoantibodies against this podocyte antigen cause ~60% of primary MGN. Anti-PLA2R IgG4 (poor classical complement activator) binds the basal surface of podocytes, complexes are shed to form subepithelial deposits, complement is activated (predominantly alternative pathway)
- Other target antigens in primary MGN: NELL1, THSD7A, EXT1/EXT2 (associated with autoimmune disease), NCAM1, Sema3B, PCDH7, HTRA1
- 20-30% is secondary (malignancy, hepatitis B, syphilis, malaria, lupus, drugs - gold, penicillamine, NSAIDs, anti-TNF agents)
Morphology:
- LM: Uniform, diffuse thickening of the glomerular capillary wall (GBM). Normal cellularity. Silver stain shows characteristic "spikes" of GBM material projecting between subepithelial deposits
- EM: Electron-dense deposits on the subepithelial (outer) surface of the GBM, with podocyte foot process effacement. Basement membrane material is deposited between deposits (spikes), eventually burying them
- IF: Granular IgG and C3 deposits along peripheral capillary loops (the classic "full-house" granular staining along GBM)
Stages (Ehrenreich-Churg):
- Stage I: Small subepithelial deposits, normal GBM
- Stage II: GBM spikes between deposits
- Stage III: Deposits enclosed within GBM
- Stage IV: Irregular thickened GBM, deposits being resorbed
Clinical Features:
- Insidious onset of nephrotic syndrome (edema, hypoalbuminemia, hyperlipidemia)
- 15% present with sub-nephrotic proteinuria
- Microscopic hematuria in up to 50%; hypertension in advanced disease
- Risk of thrombosis (especially renal vein thrombosis) is higher than in any other nephrotic syndrome
- "Rule of thirds": 1/3 spontaneous remission, 1/3 remain proteinuric but stable, 1/3 progress to ESKD
Treatment: Anti-PLA2R antibody titer guides management. Mild disease: conservative (RAAS blockade, statins). Progressive disease: immunosuppression - the Ponticelli regimen (alternating months of corticosteroids and chlorambucil/cyclophosphamide) or rituximab (now preferred first-line in many guidelines).
- Harrison's Principles of Internal Medicine 22E; Robbins, Cotran & Kumar Pathologic Basis of Disease
4. IgA Nephropathy (Berger's Disease)
Epidemiology: Most common primary GN worldwide; leading cause of renal failure globally. Particularly prevalent in Asia and among those of African ancestry carrying high-risk APOL1 alleles.
Pathogenesis: The "4-hit hypothesis":
- Elevated circulating galactose-deficient IgA-1 (Gd-IgA1) - the initiating event
- Anti-glycan autoantibodies form against the deficient hinge-region O-glycans
- Immune complexes form between Gd-IgA1 and anti-Gd-IgA1 IgG/IgA antibodies
- Complexes deposit in the mesangium, activating complement (lectin and alternative pathways) and triggering mesangial cell proliferation and injury
Morphology:
- LM: Mesangial hypercellularity and matrix expansion (focal or diffuse); may progress to segmental sclerosis or crescents
- IF: Dominant IgA deposits in the mesangium (hallmark); C3 co-deposition; IgG and IgM may also be present
- EM: Mesangial electron-dense deposits; subendothelial deposits in more severe cases
- The Oxford MEST-C score grades pathology: Mesangial hypercellularity (M), Endocapillary hypercellularity (E), Segmental sclerosis (S), Tubular atrophy/interstitial fibrosis (T), Crescents (C)
Clinical Features:
- Classic presentation: synpharyngitic (synpneumonic) gross hematuria - hematuria appearing 1-3 days into a viral URTI (distinguishes it from postinfectious GN where hematuria is 10-21 days after infection)
- Asymptomatic microscopic hematuria ± proteinuria (incidental finding)
- Proteinuria >1.5 g/g protein-to-creatinine ratio with decreased GFR indicates higher risk
- Complement levels are normal (distinguishes from PIGN)
- May progress to ESKD; patients with high-risk APOL1 alleles progress faster
Treatment: All patients need blood pressure control (target <130/80 mmHg) and maximal RAAS blockade with ACEi/ARB. Patients with proteinuria >1 g/day despite supportive care: consider systemic corticosteroids; new therapies targeting the complement system (iptacopan, sparsentan, budesonide for targeted intestinal mucosal IgA suppression) are under investigation or recently approved.
- Harrison's Principles of Internal Medicine 22E; Comprehensive Clinical Nephrology 7th Ed
5. Membranoproliferative GN (MPGN)
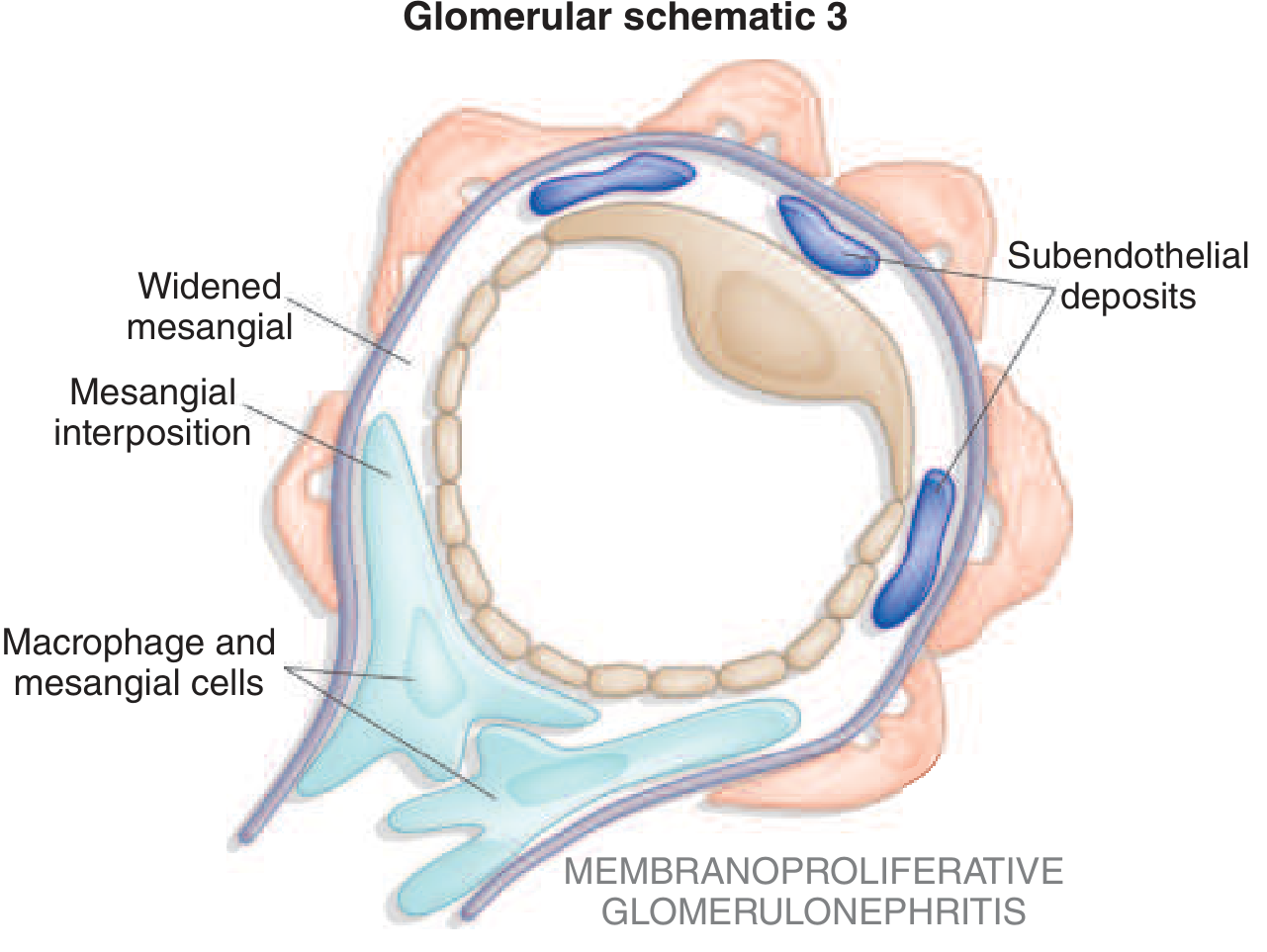
Concept: MPGN is now understood as a pattern of injury rather than a single disease. It is defined by mesangial and endocapillary hypercellularity plus GBM double-contours ("tram-tracking") due to mesangial interposition between the endothelium and GBM. It has three mechanistic subtypes:
A. Immune Complex-Mediated MPGN
Caused by chronic antigenemia with immune complex deposition:
- Infections: Hepatitis C (most common; associated with cryoglobulinemia), hepatitis B, HIV, SBE, infected shunts
- Autoimmune: SLE, Sjögren's syndrome, rheumatoid arthritis
- Monoclonal immunoglobulin deposition
- IF shows C3, IgG, IgM deposits; EM shows subendothelial ± mesangial deposits
B. Complement-Mediated MPGN (C3 Glomerulopathy)
Caused by dysregulation of the alternative complement pathway:
- Dense Deposit Disease (DDD): Formerly MPGN type II. Defined by dense osmiophilic deposits forming ribbons within the GBM on EM (pathognomonic). Primarily affects children/young adults. C3 nephritic factor (C3NeF) stabilizes the C3 convertase. Associated with partial lipodystrophy and retinal drusen. 50% progress to ESKD. Poor prognosis
- C3 Glomerulonephritis (C3GN): No dense ribbon deposits on EM but otherwise similar mechanism - mutations in complement factor H regulatory (CFHR) genes, C3NeF. Older average age (~30 years). Low serum C3 with normal C4. Staining: dominant C3 with little/no immunoglobulin on IF
- Both are managed with eculizumab (anti-C5 monoclonal antibody), steroids, and RAAS blockade
C. Monoclonal Immunoglobulin-Mediated
Seen in plasma cell dyscrasias, lymphoplasmacytic lymphoma; monoclonal light/heavy chains mediate injury
General MPGN Clinical Features:
- Presents as nephrotic syndrome, nephritic syndrome, or mixed
- Hypocomplementemia (low C3; low C4 in immune complex types)
- Indolent but progressive course; 50% develop ESKD within 10 years
- Harrison's Principles of Internal Medicine 22E; Comprehensive Clinical Nephrology 7th Ed
6. Postinfectious (Post-Streptococcal) GN
Epidemiology: Incidence has decreased markedly in developed countries due to antibiotic availability; still common in developing regions. Classic in children 5-12 years; can occur in adults.
Pathogenesis: Immune response to specific nephritogenic strains of group A beta-hemolytic streptococci (pharyngitis or skin/impetigo). Antibody-antigen immune complexes form in the circulation 10 days to 3 weeks after infection (latent period is critical - 1-3 weeks post-pharyngitis, 3-6 weeks post-impetigo). These complexes deposit subendothelially and activate complement (classical pathway), causing intense proliferative GN. Bacterial antigens involved include nephritis-associated plasmin receptor (NAPlr) and streptococcal pyrogenic exotoxin B (SpeB).
Morphology:
- LM: Diffuse endocapillary proliferation with "bloodless capillaries" - infiltration by neutrophils and monocytes fills capillary lumens; mesangial expansion
- EM: Large, hump-shaped subepithelial deposits (the "humps") - pathognomonic when present; also subendothelial deposits
- IF: Granular "starry sky" IgG and C3 along capillary walls and mesangium; the "full-house" pattern involves both C3 and IgG
Clinical Features:
- Acute nephritic syndrome: hematuria (tea- or cola-colored urine), oliguria, edema, hypertension
- Oliguria and sometimes anuria; low urine Na, concentrated urine (prerenal physiology)
- Elevated ASO titers, anti-DNase B titers, streptozyme
- Hypocomplementemia (low C3, normal C4 - alternative pathway activation) - key diagnostic finding; C3 normalizes within 8-12 weeks
- At time of presentation, infection may have resolved (no active infection)
Prognosis: Excellent in children - nearly complete recovery; adults have higher risk of persistent proteinuria, hypertension, and progression to CKD. Treatment is primarily supportive (salt/fluid restriction, antihypertensives, diuretics).
- Harrison's Principles of Internal Medicine 22E
7. Rapidly Progressive GN (RPGN) / Crescentic GN
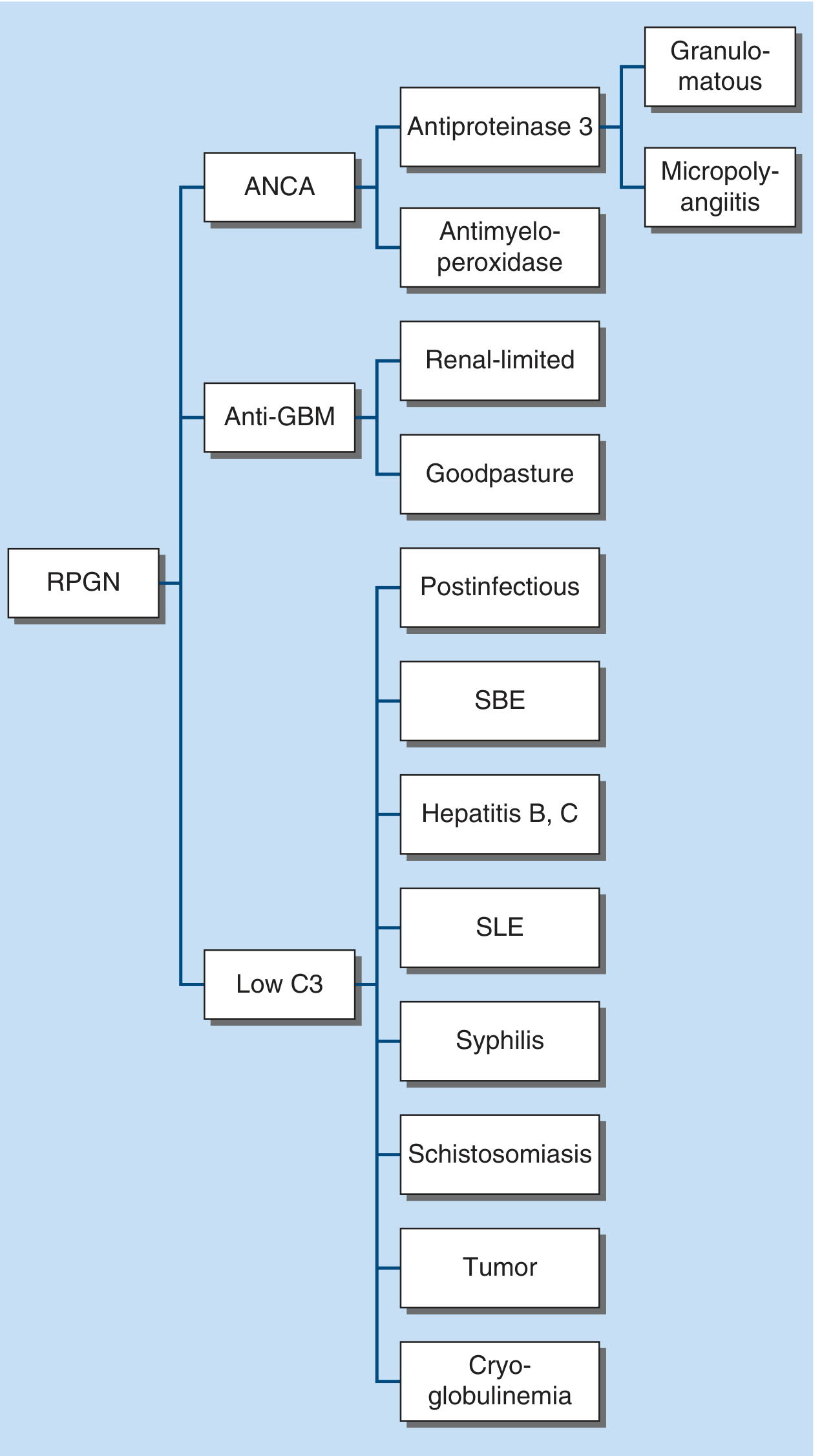
RPGN is defined clinically (rapid loss of renal function over days-weeks) and pathologically by crescents (proliferation of parietal epithelial cells and inflammatory cells surrounding and compressing glomerular capillaries) in >50% of glomeruli. There are three major immunopathologic types:
Type I: Anti-GBM Disease (Goodpasture's Disease)
- Mechanism: Autoantibodies against type IV collagen alpha-3 chain (the Goodpasture antigen) in the GBM
- IF: Linear IgG staining along GBM - pathognomonic pattern
- Clinical: When confined to kidneys only = anti-GBM nephritis; when associated with pulmonary hemorrhage = Goodpasture syndrome. Young males who smoke or have inhaled hydrocarbons are at highest risk. Fulminant presentation. Anti-GBM antibodies are diagnostic
- Treatment: Emergency plasmapheresis + cyclophosphamide/rituximab + corticosteroids. This is a single-episode disease - does not usually recur (unlike ANCA vasculitis)
Type II: Immune Complex-Mediated RPGN
- Mechanism: Immune complex deposition with complement activation (lupus nephritis, postinfectious GN, IgAN, cryoglobulinemia, Henoch-Schönlein purpura)
- Serology: Low C3 (complement consumption)
- IF: Granular deposits of IgG and complement
- Associated with: SLE, postinfectious GN, hepatitis B/C, SBE, cryoglobulinemia
Type III: Pauci-Immune RPGN (ANCA-Associated)
- Mechanism: ANCA (antineutrophil cytoplasmic antibodies) target neutrophil granule proteins - most destructive form
- c-ANCA (anti-PR3): Granulomatosis with polyangiitis (formerly Wegener's)
- p-ANCA (anti-MPO): Microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
- IF: Sparse or absent deposits ("pauci-immune") - distinguishes from Types I and II
- Clinical: Can present with pulmonary-renal syndrome (hemoptysis, epistaxis, sinusitis, nasal congestion). ANCA vasculitides may recur over time
- Treatment: Induction with cyclophosphamide or rituximab + high-dose steroids; maintenance with rituximab or azathioprine; anti-complement therapy (avacopan) is now incorporated in guidelines
Note: Sometimes both anti-GBM and ANCA are simultaneously positive ("double-positive disease"), which has intermediate prognosis.
- Harrison's Principles of Internal Medicine 22E
8. Fibrillary GN and Immunotactoid GN
These are rare, distinct entities with organized deposits:
- Fibrillary GN: Glomerular deposits composed of randomly arranged fibrils 10-30 nm in diameter (larger than amyloid fibrils of ~10 nm), non-congophilic (Congo red negative). Often associated with autoimmune disease or dysproteinemia
- Immunotactoid GN: Organized microtubular deposits >30 nm, often with parallel arrangement, almost always associated with monoclonal gammopathy or lymphoplasmacytic lymphoma
Both present with proteinuria (often nephrotic range), hematuria, and hypertension, and have a poor prognosis.
- Brenner and Rector's The Kidney
Summary Table
| Type | Primary Syndrome | Key Pathology | Hallmark Finding | Complement | Treatment |
|---|---|---|---|---|---|
| Minimal Change Disease | Nephrotic | Foot process effacement on EM; normal LM | Anti-nephrin antibodies; normal LM | Normal | Corticosteroids |
| FSGS | Nephrotic | Focal/segmental sclerosis; podocytopathy | Non-selective proteinuria; poor steroid response | Normal | Steroids, CNI; treat underlying cause |
| Membranous Nephropathy | Nephrotic | Subepithelial deposits; GBM spikes on silver stain | Anti-PLA2R antibodies; granular IF on GBM | Normal | Rituximab; Ponticelli regimen |
| IgA Nephropathy | Hematuria ± nephritic/nephrotic | Mesangial IgA deposits | Synpharyngitic hematuria; mesangial IgA on IF | Normal | RAAS blockade; steroids if proteinuria >1 g/day |
| MPGN / C3G | Mixed nephritic-nephrotic | Double-contour GBM; mesangial proliferation | Tram-tracking; low C3 | Low C3 | Treat underlying cause; eculizumab for C3G |
| Postinfectious GN | Acute nephritic | Subepithelial humps on EM; endocapillary proliferation | Low C3; elevated ASO titers | Low C3 | Supportive; antihypertensives |
| RPGN (Crescentic) | Rapidly progressive | Crescents >50% glomeruli | Type I: linear IF; Type II: granular IF; Type III: pauci-immune IF | Low C3 (Types I, II) | Plasmapheresis; cyclophosphamide/rituximab |
| Fibrillary/Immunotactoid | Nephrotic/mixed | Organized fibrillar deposits on EM; Congo red negative | Microtubular deposits >10 nm; non-amyloid | Variable | Immunosuppression; treat dysproteinemia |
Sources: Harrison's Principles of Internal Medicine 22E (2025); Robbins, Cotran & Kumar Pathologic Basis of Disease; Comprehensive Clinical Nephrology 7th Ed; Brenner and Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.