Bronchiectasis definition
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Bronchiectasis - Definition
Bronchiectasis is the permanent, abnormal dilation of bronchi and bronchioles caused by destruction of the smooth muscle and supporting elastic tissue of the bronchial wall. It is characteristically irreversible and almost always occurs secondary to an underlying condition - it is not a primary disorder.
Core Definition (from Authoritative Sources)
"Bronchiectasis is the permanent dilation of bronchi and bronchioles caused by destruction of smooth muscle and supporting elastic tissue; it typically results from or is associated with chronic necrotizing infections."
- Robbins & Kumar Basic Pathology
"In bronchiectasis, there is, by definition, an abnormal and permanent dilatation of the bronchi; this is usually present to a much greater degree than is found in COPD and is often accompanied by airway distortion."
- Murray & Nadel's Textbook of Respiratory Medicine
"In bronchiectasis, there is, by definition, an abnormal and permanent dilatation of the bronchi... often accompanied by airway distortion. There is exaggeration of the muscular ridges and the presence of multiple bronchial gland-based pits. The large airway walls can be thickened and/or irregularly thinned as a result of inflammation and fibrosis, and there is often inspissated mucus or actual purulent material."
- Murray & Nadel's Textbook of Respiratory Medicine
Key Characteristics
| Feature | Detail |
|---|---|
| Nature | Permanent (irreversible) |
| Pathology | Dilation + destruction of bronchial wall (smooth muscle + elastic tissue) |
| Primary vs. Secondary | Always secondary to infection, obstruction, or underlying disease |
| Pathogenesis | Two intertwined processes: obstruction and chronic infection |
| Airways affected | Bronchi and bronchioles; typically distal airways, lower lobes bilaterally |
| Degree of dilation | Up to 4 times normal diameter |
Pathogenesis in Brief
Two processes drive bronchiectasis in a vicious cycle:
- Obstruction (foreign body, mucus, tumor) impairs secretion clearance, allowing infection to take hold
- Chronic necrotizing infection damages the bronchial wall, causes peribronchial fibrosis, and progressively distends the airways
Either can be the initiating event - once established, they perpetuate each other.
Classic Clinical Features
- Chronic cough producing copious purulent sputum (typically >30 mL/day)
- Hemoptysis
- Recurrent pulmonary infections
- Common pathogens: Haemophilus influenzae, Staphylococcus aureus, Pseudomonas aeruginosa
Common Predisposing Conditions
- Bronchial obstruction (tumor, foreign body, mucus impaction)
- Cystic fibrosis (most common cause of severe widespread bronchiectasis)
- Immunodeficiency states (especially immunoglobulin deficiencies)
- Primary ciliary dyskinesia (immobile cilia syndrome)
- Necrotizing/suppurative pneumonia (S. aureus, Klebsiella)
- Post-tuberculosis (significant in endemic areas)
- Post-COVID-19 pneumonia (increasingly reported)
Sources: Robbins & Kumar Basic Pathology; Murray & Nadel's Textbook of Respiratory Medicine; Fishman's Pulmonary Diseases and Disorders
Nephro toxic antibiotics
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
Here is a comprehensive overview of nephrotoxic antibiotics, drawn from authoritative nephrology and pharmacology textbooks.
Nephrotoxic Antibiotics
The kidney is highly susceptible to drug toxicity due to its extreme blood perfusion and its role in concentrating filtered substances. The diagram below shows the full spectrum of nephrotoxic mechanisms:
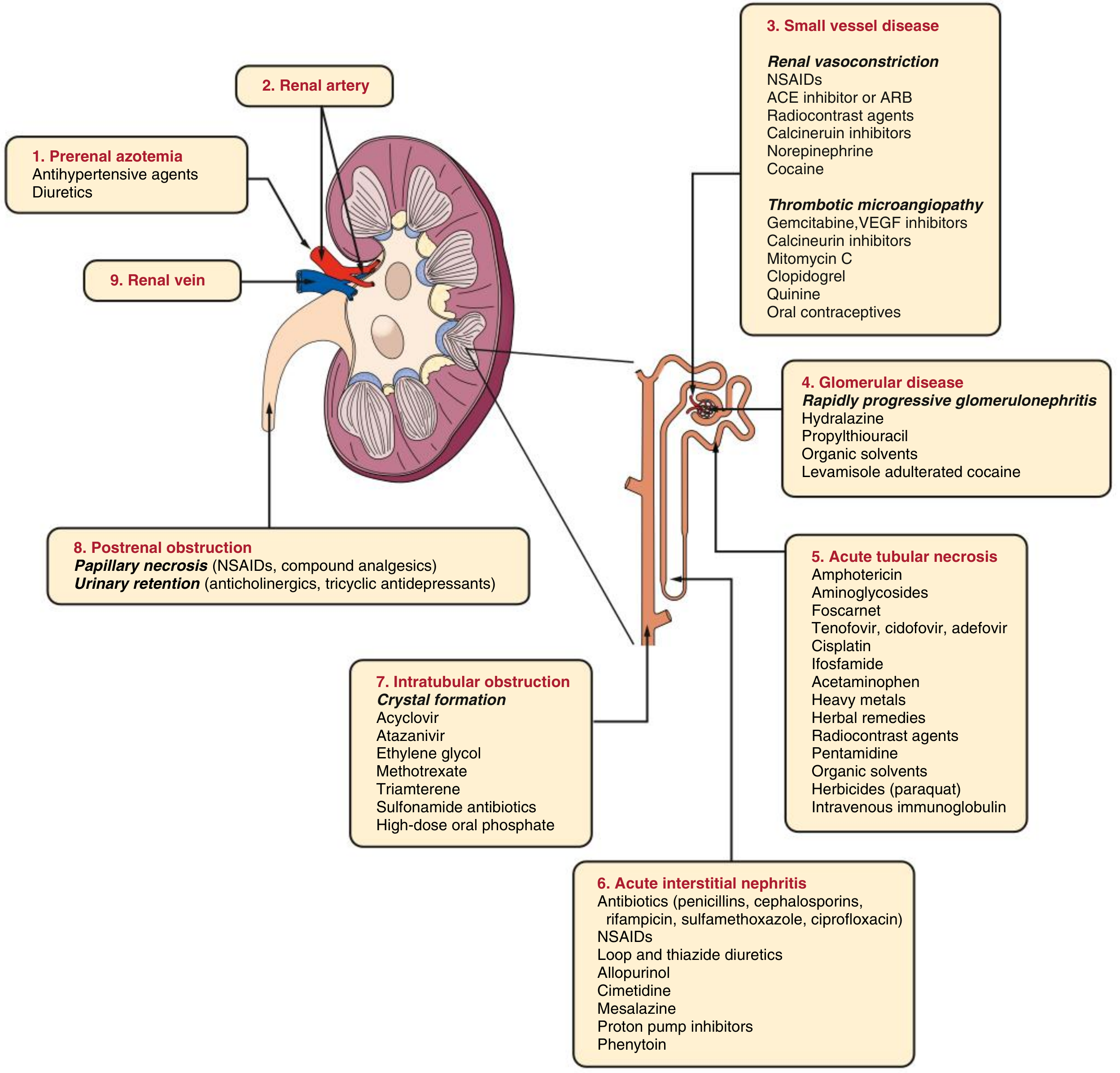
1. Aminoglycosides (Most Common Antibiotic Nephrotoxin)
Drugs: Gentamicin, tobramycin, amikacin, netilmicin, plazomicin
Mechanism:
- Freely filtered at the glomerulus and accumulate in proximal tubular cells (cortex) via megalin-mediated endocytosis
- Cause formation of myeloid bodies (membrane fragments in lysosomes) from inhibition of lysosomal enzymes
- Lead to oxidative stress, disruption of subcellular organelles, and mitochondrial dysfunction
- Result in acute tubular necrosis (ATN)
Clinical features:
- Nonoliguric AKI in 10-30% of courses (even with therapeutic levels)
- Rising serum creatinine after 5-7 days of therapy
- Low-grade proteinuria, renal tubular epithelial (RTE) cells and granular casts in urine before clinical AKI
- Hypomagnesemia, hypokalemia (tubular wasting)
- In severe cases: Fanconi syndrome or Bartter-like syndrome
Risk factors for aminoglycoside nephrotoxicity:
| Risk Factor |
|---|
| Prolonged therapy (>10 days) |
| Volume depletion |
| Sepsis |
| Pre-existing kidney disease |
| Hypokalemia |
| Advanced age |
| Combination with cephalosporins (especially cephalothin) |
| Co-administration with vancomycin, amphotericin B, cisplatin |
Prevention: Single daily dosing (tubular absorption is saturable); monitor drug levels every 2-3 days; maintain hydration. Amikacin and plazomicin may be less nephrotoxic than gentamicin.
2. Vancomycin
Mechanism:
- Excreted by glomerular filtration; accumulates in proximal tubular cells via basolateral secretion
- High-dose causes oxidative stress and triggers intrarenal apoptotic pathways
- ATN is the predominant lesion; rarely causes acute interstitial nephritis (AIN) or DRESS syndrome
- Can crystallize in tubules and cause intratubular obstruction
Clinical features:
- AKI associated with trough levels >15 µg/mL (dose-dependent)
- Co-administration with piperacillin-tazobactam significantly increases AKI risk
- Generally reversible; high-flux hemodialysis can remove drug if levels are very high
Risk factors: Total dose >4 g/day, long duration, critical illness, concurrent nephrotoxins or diuretics
3. Amphotericin B (Antifungal Antibiotic)
Mechanism:
- Binds to cholesterol in tubular cell membranes (and ergosterol in fungal membranes), creating aqueous pores
- Sodium influx increases Na⁺/K⁺-ATPase activity and depletes cellular energy
- Causes renal vasoconstriction via tubuloglomerular feedback
- Direct tubular toxicity via reactive oxygen species
Clinical features:
- Degree of injury proportional to cumulative dose (usually after >2-3 g)
- Polyuria (loss of concentrating ability), hypomagnesemia, hypokalemia, hypocalcemia
- Distal renal tubular acidosis (non-anion gap metabolic acidosis)
- Nonoliguric, progressive AKI
Prevention: Saline loading (volume expansion), liposomal amphotericin B reduces AKI risk by ~50%
4. Polymyxins (Colistin / Polymyxin B)
Mechanism: Cationic detergent action disrupts bacterial and renal tubular cell membrane integrity
Clinical features:
- Dose-dependent, reversible nephrotoxicity - the most important adverse effect
- Also causes neurotoxicity (paresthesias, muscle weakness) but less commonly
Use context: Reserved for multidrug-resistant (MDR) gram-negative infections (carbapenem-resistant Acinetobacter, Pseudomonas)
5. Antibiotics Causing Acute Interstitial Nephritis (AIN)
These antibiotics trigger an immune/allergic reaction in the renal interstitium:
| Antibiotic Class | Examples |
|---|---|
| Beta-lactams | Penicillins, cephalosporins, carbapenems |
| Rifampicin | - |
| Sulfonamides | Sulfamethoxazole (in TMP-SMX) |
| Fluoroquinolones | Ciprofloxacin |
Features: Fever, rash, eosinophilia ("classic triad" in <30%), eosinophiluria, sterile pyuria; may require steroid treatment.
6. Antibiotics Causing Crystal Nephropathy / Intratubular Obstruction
| Drug | Mechanism |
|---|---|
| Sulfonamides | Crystal precipitation in tubules |
| Acyclovir (antiviral) | Birefringent needle-shaped crystals (esp. IV bolus) |
| Ciprofloxacin | Crystal deposition in acid urine |
7. Other Antimicrobials
| Drug | Mechanism | Notes |
|---|---|---|
| Foscarnet | Direct tubular toxicity | Used in CMV/HSV |
| Pentamidine | Tubular toxicity | Used in PCP |
| Tenofovir (TDF) | Mitochondrial toxicity, proximal tubulopathy | Can cause Fanconi syndrome; TAF (newer formulation) is less nephrotoxic |
| Cidofovir | Proximal tubular toxicity | High nephrotoxicity; requires probenecid prophylaxis |
Summary Table
| Antibiotic | Primary Mechanism | Lesion |
|---|---|---|
| Aminoglycosides | Proximal tubular accumulation, oxidative stress | ATN |
| Vancomycin | Tubular apoptosis, crystal obstruction | ATN, AIN |
| Amphotericin B | Membrane pore formation + vasoconstriction | ATN + RTA |
| Polymyxins/Colistin | Membrane disruption | Tubular toxicity |
| Penicillins, cephalosporins, rifampicin, sulfonamides, fluoroquinolones | Immune/allergic | AIN |
| Sulfonamides, acyclovir | Crystal precipitation | Intratubular obstruction |
| Tenofovir, cidofovir, foscarnet | Mitochondrial/tubular toxicity | Proximal tubulopathy |
Sources: Comprehensive Clinical Nephrology 7e; National Kidney Foundation Primer on Kidney Diseases 8e; Harrison's Principles of Internal Medicine 22e; Goodman & Gilman's Pharmacological Basis of Therapeutics
Hepatorenal syndrome
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Excellent - I now have a comprehensive set of data. Here is the full answer:
Hepatorenal Syndrome (HRS)
Definition
HRS is a functional form of acute kidney injury (AKI) without intrinsic renal pathology, occurring in patients with advanced cirrhosis, acute liver failure, or severe alcoholic hepatitis. Histologically, the kidneys are normal - the syndrome results entirely from hemodynamic derangement. Kidneys from HRS patients have even been successfully transplanted into non-cirrhotic recipients with full recovery of function.
- Occurs in ~10% of patients with advanced cirrhosis or acute liver failure
- AKI occurs in 15-25% of all hospitalized cirrhotic patients; HRS accounts for 10-30% of these
- Annual frequency of HRS in cirrhotic patients with ascites: ~8% (up to 40% in some reports)
Pathophysiology
The pathophysiology involves three intertwined components:
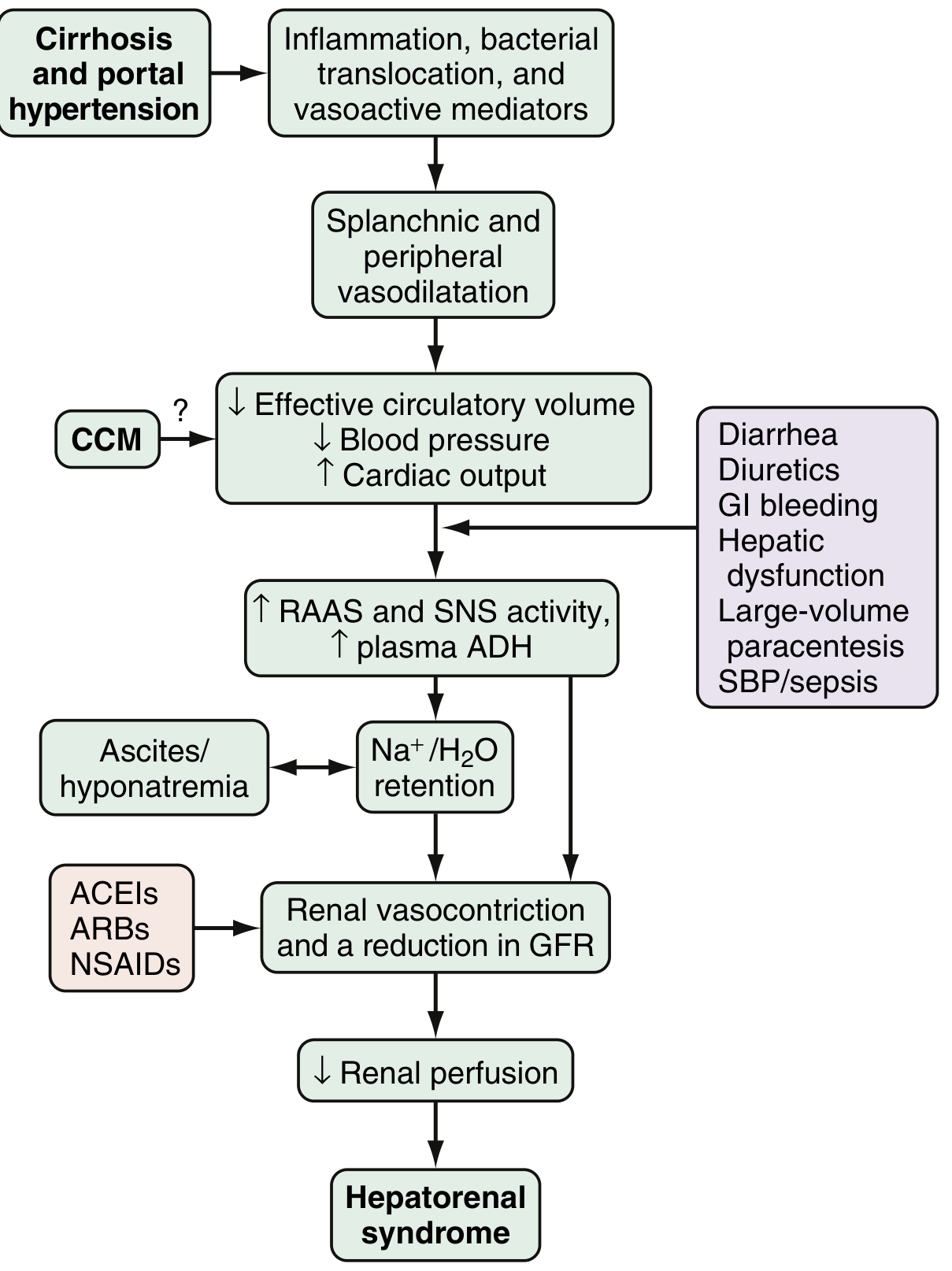
1. Splanchnic and Systemic Arterial Vasodilation
- Portal hypertension triggers release of vasoactive mediators: nitric oxide (NO), carbon monoxide, glucagon, prostacyclin, adrenomedullin, endogenous opiates
- These cause marked splanchnic and peripheral vasodilation
- Results in decreased effective circulating blood volume and reduced blood pressure
- Early compensation: increased heart rate and cardiac output (hyperdynamic circulation)
2. Compensatory Renal Vasoconstriction
- As splanchnic vasodilation progresses beyond compensation, the body activates:
- Renin-angiotensin-aldosterone system (RAAS)
- Sympathetic nervous system (SNS)
- Non-osmotic ADH release
- These responses cause intense renal vasoconstriction - a shift in the balance between systemic vasodilation and renal vasoconstriction
- Leads to increased renal vascular resistance, decreased renal perfusion, and reduced GFR
- Mediators involved: endothelins, altered prostaglandins, kallikreins, F2-isoprostanes
- Result: Na⁺/H₂O retention → ascites and hyponatremia
3. Cardiac Dysfunction (Cirrhotic Cardiomyopathy - CCM)
- Impaired cardiac output further worsens renal hypoperfusion
- Patients who develop HRS have lower cardiac output and higher circulating norepinephrine/renin than those who do not
Key precipitants: SBP/sepsis (30%), severe alcoholic hepatitis (25%), large-volume paracentesis without albumin (10%), GI bleeding, over-diuresis
Types of HRS (Updated Nomenclature)
| Old Term | New Term | Characteristics |
|---|---|---|
| Type 1 HRS | HRS-AKI | Rapid, progressive AKI (stage 2-3); serum creatinine doubles to >2.5 mg/dL within 2 weeks; poor prognosis |
| Type 2 HRS | HRS-CKD | Stable, moderate reduction in GFR; gradual course; associated with refractory ascites; better prognosis than Type 1 |
Diagnostic Criteria (International Club of Ascites, 2015)
All of the following must be present:
- Cirrhosis with ascites
- AKI by ICA criteria: rise in serum creatinine ≥0.3 mg/dL within 48 h OR ≥50% rise from baseline within 7 days
- No response after ≥48 h of diuretic withdrawal + IV albumin (1 g/kg/day, max 100 g/day)
- Absence of shock
- No recent nephrotoxic drugs (NSAIDs, ACEIs, ARBs, certain antibiotics)
- No intrinsic renal disease: proteinuria <500 mg/day, no microhematuria (>50 RBC/hpf), normal renal ultrasound
Note: HRS is a diagnosis of exclusion - ATN, AIN, glomerulonephritis, and obstructive uropathy must all be ruled out.
Clinical Features
- Most patients are asymptomatic except for decreased urine output
- Oliguria (urine output often <500 mL/day in HRS-AKI)
- Urinary sodium typically very low (<10 mEq/L) - reflects avid sodium retention
- Urine osmolality > plasma osmolality
- Rising serum creatinine and BUN
- Hepatic encephalopathy may be the presenting feature
- Background: jaundice, ascites, spider angiomata, asterixis
Management
Prevention (key):
- Avoid volume depletion (over-diuresis, lactulose diarrhea, large-volume paracentesis without albumin)
- Avoid nephrotoxins (NSAIDs, ACEIs, ARBs, certain antibiotics)
- Treat infections promptly (especially SBP)
- IV albumin 1.5 g/kg on day 1 + 1 g/kg on day 3 for SBP patients (prevents HRS)
- Beta-blockers + band ligation to prevent variceal bleeding
Treatment of established HRS:
| Intervention | Details |
|---|---|
| Stop nephrotoxins + diuretics | First step |
| IV Albumin | 1 g/kg/day bolus (max 100 g/day); then 20-60 g/day to maintain CVP 10-15 cmH₂O |
| Terlipressin (1st line vasoconstrictor) | 1 mg IV q4h → up to 2 mg IV q4h if creatinine doesn't improve 25% by day 3 |
| Norepinephrine (ICU alternative) | 0.1-0.7 µg/kg/min IV; titrate to MAP increase ≥10 mmHg |
| Midodrine + Octreotide + Albumin | Midodrine 2.5-15 mg PO TID; Octreotide 100-200 µg SC TID; 3rd line in the US |
| TIPS | Useful in HRS-CKD / refractory ascites; benefits mainly type 2; risk of worsening hepatic function |
| Renal replacement therapy | Bridge to transplant if vasoconstrictors fail |
| Liver transplantation | Definitive treatment - reverses HRS in 83%; 3-year survival ~60% (vs 70-80% without HRS) |
Duration of vasoconstrictor therapy: maximum 2 weeks, until HRS reverses or liver transplant is performed.
Prognosis
- Without treatment: median survival of Type 1 HRS is 2 weeks
- With terlipressin + albumin: reversal in ~40-50% of cases
- Liver transplantation is the only modality that can reverse both liver dysfunction and HRS; 35% still require long-term renal replacement therapy post-transplant
Sources: Sleisenger & Fordtran's Gastrointestinal and Liver Disease; Harrison's Principles of Internal Medicine 22e; Comprehensive Clinical Nephrology 7e
This is a shared conversation. Sign in to Orris to start your own chat.