Loop diuretics
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive material from multiple major pharmacology references. Here is a complete review of loop diuretics:
Loop Diuretics
Overview
Loop diuretics are the most potent class of diuretics available, capable of inducing a brisk natriuresis even when renal function is significantly compromised. They are sometimes called high-ceiling diuretics because their dose-response curve reaches a substantially higher maximum than other diuretic classes. The four agents available are furosemide, bumetanide, torsemide, and ethacrynic acid.
Chemical Classification
| Drug | Chemical Class | Sulfonamide Moiety |
|---|---|---|
| Furosemide | Sulfonamide-based | Yes |
| Bumetanide | Sulfonamide-based | Yes |
| Torsemide | Sulfonylurea | Yes |
| Ethacrynic acid | Phenoxyacetic acid derivative | No |
Ethacrynic acid is the only loop diuretic without a sulfonamide moiety, making it the agent of choice in patients with a confirmed sulfa allergy. Its reactive methylene group forms a covalent adduct with cysteine's free sulfhydryl group, and this cysteine adduct is the active form of the drug.
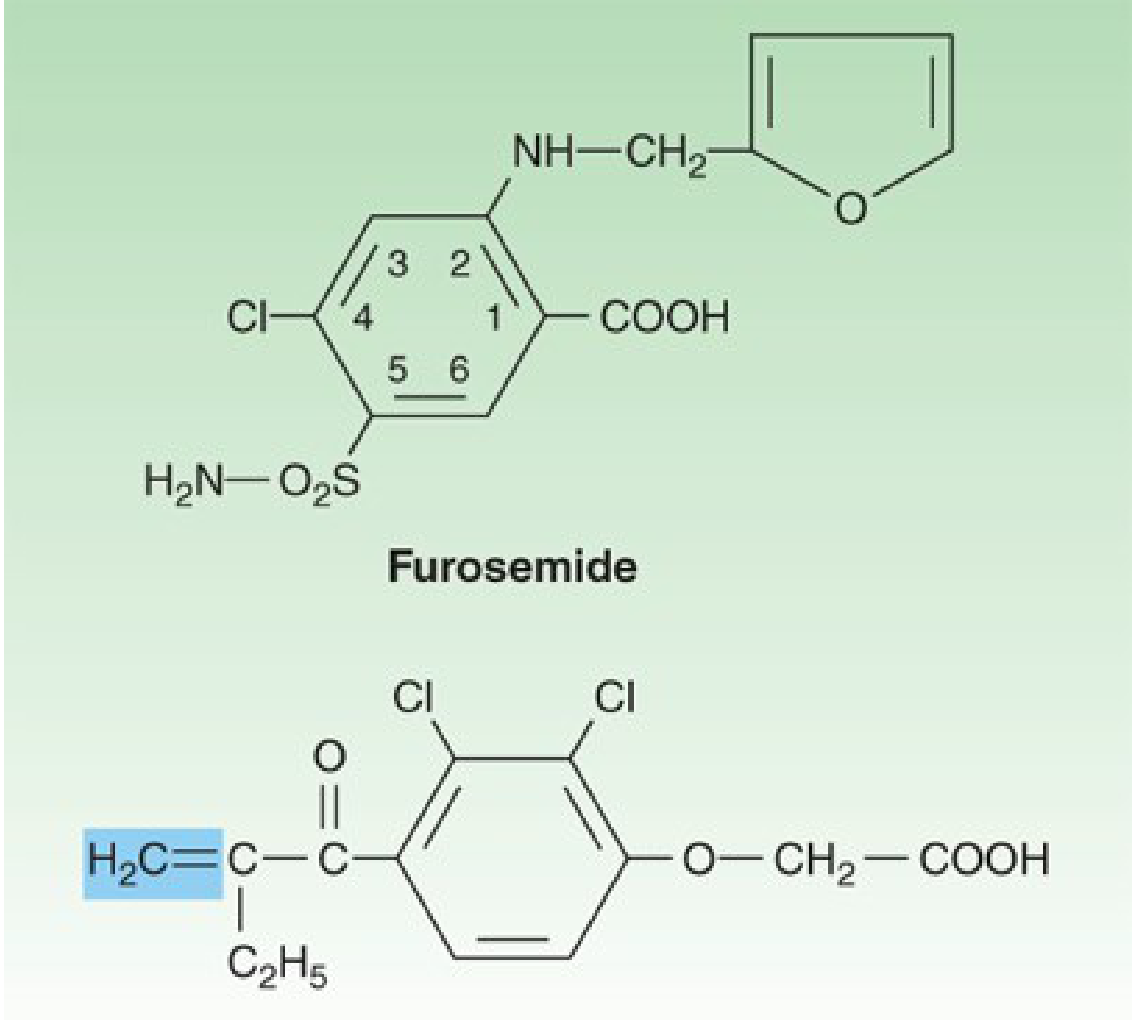
Furosemide (top) and ethacrynic acid (bottom). The shaded methylene group on ethacrynic acid is reactive and combines with sulfhydryl groups. - Katzung's Basic and Clinical Pharmacology, 16th Ed.
Site and Mechanism of Action
Loop diuretics act primarily in the thick ascending limb (TAL) of the loop of Henle, where they bind to and inhibit the Na⁺-K⁺-2Cl⁻ cotransporter (NKCC2/SLC12A1) on the luminal (apical) membrane of tubular epithelial cells.
Why is this site so effective?
- The TAL normally reabsorbs ~25% of the filtered Na⁺ load
- Nephron segments downstream of the TAL lack the reabsorptive capacity to compensate for the massive salt delivery escaping the TAL
- The proximal tubule, by contrast, reabsorbs ~65% of filtered Na⁺, but diuretics acting only there have limited efficacy because the TAL rescues most of the escaped load
Secondary consequences of NKCC2 inhibition:
- Reduced lumen-positive transepithelial potential (normally generated by K⁺ recycling back into the lumen) - this potential is the driving force for divalent cation reabsorption, so blocking it increases Mg²⁺ and Ca²⁺ excretion
- Disruption of the hypertonic medullary interstitium, impairing water reabsorption in the collecting duct even in the presence of AVP (antidiuretic hormone) - resulting in nearly isotonic urine
- Increased Na⁺ and water delivery to the distal nephron markedly enhances K⁺ excretion, especially in the presence of elevated aldosterone levels
Prostaglandin effect: Loop diuretics induce renal COX-2 expression, increasing prostaglandin (particularly PGE₂) synthesis. PGE₂ itself inhibits salt transport in the TAL, contributing to the diuretic response. This explains why NSAIDs (which inhibit COX-2) can blunt the response to loop diuretics.
- Goodman & Gilman's The Pharmacological Basis of Therapeutics
- Katzung's Basic and Clinical Pharmacology, 16th Ed.
- National Kidney Foundation Primer on Kidney Diseases, 8th Ed.
Pharmacokinetics
| Drug | Oral Bioavailability | t½ | Elimination | Onset (oral) |
|---|---|---|---|---|
| Furosemide | ~60% (variable: 12-112%) | ~1.5 h | ~65% renal, ~35% hepatic | 2-3 hours |
| Bumetanide | ~80% | ~0.8 h | ~62% renal, ~38% hepatic | Similar to torsemide |
| Torsemide | ~80-90% (consistent) | longer | Predominantly hepatic | ~1 hour |
| Ethacrynic acid | ~100% | ~1 h | ~67% renal, ~33% hepatic | - |
Key pharmacokinetic points:
-
All loop diuretics are highly protein-bound (mainly albumin) and therefore reach the tubular lumen primarily by active tubular secretion via organic anion transporters (OAT1/OAT2) at the basolateral membrane of the proximal tubule - NOT by glomerular filtration
-
Tubular secretion is impaired by: endogenous organic acids (accumulate in CKD and uremia), NSAIDs, probenecid, and salicylates - all competing for the same transporter
-
Furosemide has the most variable bioavailability - the coefficient of variation is 25-43%; switching formulations does not standardize response
-
Torsemide has more consistent absorption and longer duration (4-6 hours vs. 2-3 hours for furosemide), with at least one active metabolite
-
In CKD: furosemide pharmacokinetics are most significantly altered; bumetanide and torsemide (both hepatically metabolized) are less affected
-
National Kidney Foundation Primer on Kidney Diseases, 8th Ed.
-
Katzung's Basic and Clinical Pharmacology, 16th Ed.
-
Goodman & Gilman's
Relative Potency
| Drug | Equivalent Dose |
|---|---|
| Furosemide | 20 mg (or 40 mg by some references) |
| Torsemide | 10 mg |
| Bumetanide | 0.5 mg |
| Ethacrynic acid | ~50 mg |
Bumetanide is approximately 40x more potent than furosemide by weight.
Hemodynamic Effects
Beyond diuresis, loop diuretics have direct vascular effects:
-
Furosemide acts as a venodilator and reduces right atrial and pulmonary capillary wedge pressure within minutes of IV administration - before measurable diuresis occurs
-
This effect is prostaglandin-mediated and is blocked by indomethacin
-
Furosemide also increases renal blood flow via prostaglandin-mediated vasodilation of renal vasculature
-
There may be a transient rise in systemic vascular resistance due to renin secretion stimulation at the macula densa (RAS activation) - this reinforces the importance of combining loop diuretics with vasodilators in acute pulmonary edema with preserved BP
-
Braunwald's Heart Disease
Clinical Uses
| Indication | Notes |
|---|---|
| Acute pulmonary edema | First-line; rapid venodilation + natriuresis reduces LV filling pressures |
| Chronic heart failure | Reduces mortality, hospitalizations, worsening HF; torsemide may be preferred over furosemide due to more reliable absorption |
| Hypertension (resistant or low GFR <30 mL/min) | Loop diuretics preferred; NOT first-line in normal renal function (thiazides are more effective there) |
| Nephrotic syndrome edema | Often refractory to other diuretics; loop agents may be the only option |
| Hepatic cirrhosis with ascites | Used with caution to avoid volume contraction |
| Hypercalcemia | Combined with isotonic saline to prevent volume depletion; increases Ca²⁺ excretion by 30% |
| Hyperkalemia | Increases urinary K⁺ excretion; combined with IV NaCl if patient is normo-/hypovolemic |
| Life-threatening hyponatremia | Combined with hypertonic saline; loop diuretics block urine concentration |
| CKD-related edema | Higher doses needed due to right-shifted dose-response curve |
| Drug overdose (forced diuresis) | Facilitates renal elimination of offending drug |
| Acute renal failure | Listed as an indication (to increase urine output) |
- Goodman & Gilman's; Katzung's
Adverse Effects and Toxicity
Electrolyte and Fluid Disorders:
- Hyponatremia - from overzealous use
- Hypokalemia - increased distal Na⁺ delivery + aldosterone activation; can precipitate arrhythmias (especially with digoxin)
- Hypomagnesemia - K⁺ and Mg²⁺ losses often occur together; a risk factor for arrhythmias
- Hypochloremic metabolic alkalosis - increased urinary H⁺ and Cl⁻ loss
- Hypocalcemia (rare) - can cause tetany in severe cases; loop diuretics should be avoided in postmenopausal osteopenic women
- Volume depletion - hypotension, reduced GFR, circulatory collapse, thromboembolic events
- Hepatic encephalopathy - in patients with liver disease, volume depletion can precipitate this
Ototoxicity:
- Tinnitus, hearing impairment (usually reversible), deafness, vertigo, sense of ear fullness
- Most common with rapid IV administration; least with oral dosing
- Ethacrynic acid has the highest ototoxicity risk among loop diuretics and is reserved for patients who cannot tolerate others
- Risk is additive with aminoglycosides, cisplatin, carboplatin, paclitaxel
Hyperuricemia: Initial increase then longer-lived decrease in uric acid excretion; can precipitate gout
Hyperglycemia: Especially with concomitant sulfonylureas
Drug Interactions
| Interacting Drug | Effect |
|---|---|
| NSAIDs / probenecid | Blunted diuretic response (compete for proximal tubule secretion; NSAIDs also reduce PGE₂) |
| Aminoglycosides, cisplatin, carboplatin, paclitaxel | Synergistic ototoxicity |
| Digoxin | Hypokalemia increases risk of digoxin-induced arrhythmias |
| Lithium | Increased plasma lithium levels |
| Propranolol | Increased propranolol plasma levels |
| Sulfonylureas | Hyperglycemia |
| Amphotericin B | Increased nephrotoxicity and electrolyte imbalances |
| Thiazide diuretics | Synergistic diuresis (can cause profound diuresis) - used therapeutically in diuretic resistance |
Diuretic Resistance
The dose-response curve is sigmoidal. It can be shifted downward and rightward (requiring higher doses for same effect) in:
- Extracellular volume depletion
- Heart failure
- Nephrotic syndrome
- CKD (organic acid accumulation impairs tubular secretion)
- NSAID use
- Reduced protein binding (hypoalbuminemia alters pharmacokinetics)
Strategies for resistance: Increasing dose, switching to torsemide (more consistent absorption), adding a thiazide (metolazone + furosemide combination is classic), or IV administration.
Summary Comparison
| Feature | Furosemide | Bumetanide | Torsemide | Ethacrynic acid |
|---|---|---|---|---|
| Sulfonamide | Yes | Yes | Yes | No |
| Bioavailability | Variable (12-112%) | ~80% | ~80-90% | ~100% |
| Potency | 1x | ~40x | ~2x | 0.7x |
| Duration | 2-3 h | ~2 h | 4-6 h | ~2 h |
| Ototoxicity | + | + | + | +++ (highest) |
| Best use in sulfa allergy | No | No | No | Yes |
Sources: Goodman & Gilman's The Pharmacological Basis of Therapeutics; Katzung's Basic and Clinical Pharmacology 16th Ed.; National Kidney Foundation Primer on Kidney Diseases 8th Ed.; Braunwald's Heart Disease; Goldman-Cecil Medicine
Raas
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Excellent - I now have very rich material from multiple authoritative sources. Here is a comprehensive review:
Renin-Angiotensin-Aldosterone System (RAAS)
Overview
The RAAS is the principal hormonal cascade regulating extracellular fluid volume, sodium balance, and blood pressure. It consists of a chain of enzymatic steps - from renin release to angiotensin II (Ang II) generation to aldosterone secretion - that collectively drive vasoconstriction and sodium retention to restore perfusion pressure when it falls.
The Cascade: Step by Step
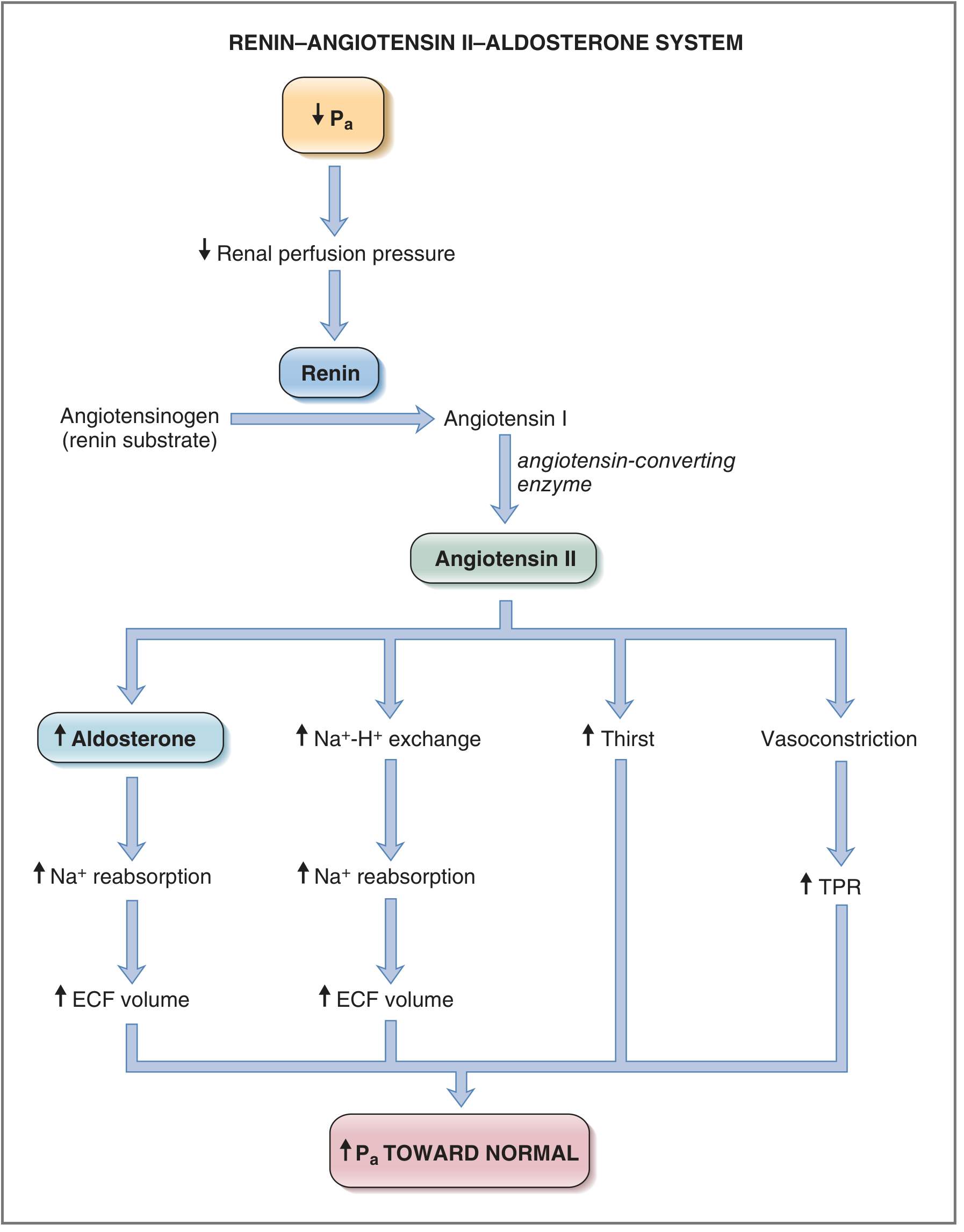
Fig. 4.33 - Renin-Angiotensin-Aldosterone System (Costanzo Physiology, 7th Ed.)
Step 1 - Renin
Source: Juxtaglomerular (JG) cells (granular cells) in the wall of the afferent arteriole of the kidney, within the juxtaglomerular apparatus (JGA).
Synthesis: Produced as prepro-renin → pro-renin → active renin (340 amino acid glycoprotein) by cleavage of a 43-amino acid N-terminal prosegment.
- Prorenin circulates at 80-90% of total; active renin is released by exocytosis on stimulation
- Extrarenal tissues (adrenal, ovaries, testes, placenta, retina) also produce prorenin
- Plasma renin disappears entirely after nephrectomy
Function: Cleaves angiotensinogen (a liver-derived glycoprotein, MW ~57,000) at the N-terminal to release the decapeptide Angiotensin I (Ang I).
Step 2 - Angiotensin I
- A decapeptide with little or no intrinsic biologic activity
- Serves solely as substrate for ACE
- Can also be converted by aminopeptidases to [des-Asp]-Ang I, then to Ang III by ACE
Step 3 - ACE and Angiotensin II
ACE (Angiotensin-Converting Enzyme, Kininase II):
- A dipeptidyl carboxypeptidase located on the luminal surface of vascular endothelial cells throughout the body (especially pulmonary vasculature)
- Cleaves a dipeptide from the C-terminus of Ang I → forms the octapeptide Angiotensin II (Ang II)
- Also cleaves (inactivates) bradykinin - this is why ACE inhibitors cause bradykinin accumulation and a dry cough
- Cannot cleave substrates with a penultimate prolyl residue (so Ang II is not further degraded by ACE)
ACE2 (a homolog):
- Carboxypeptidase (removes a single C-terminal amino acid)
- Converts Ang I → Ang 1-9 and Ang II → Ang 1-7
- Does NOT hydrolyze bradykinin; NOT inhibited by ACE inhibitors
- Ang 1-7 acts on the Mas receptor → vasodilation, natriuresis, anti-proliferative effects (counter-regulatory arm)
- ACE2 is also the entry receptor for SARS-CoV-2
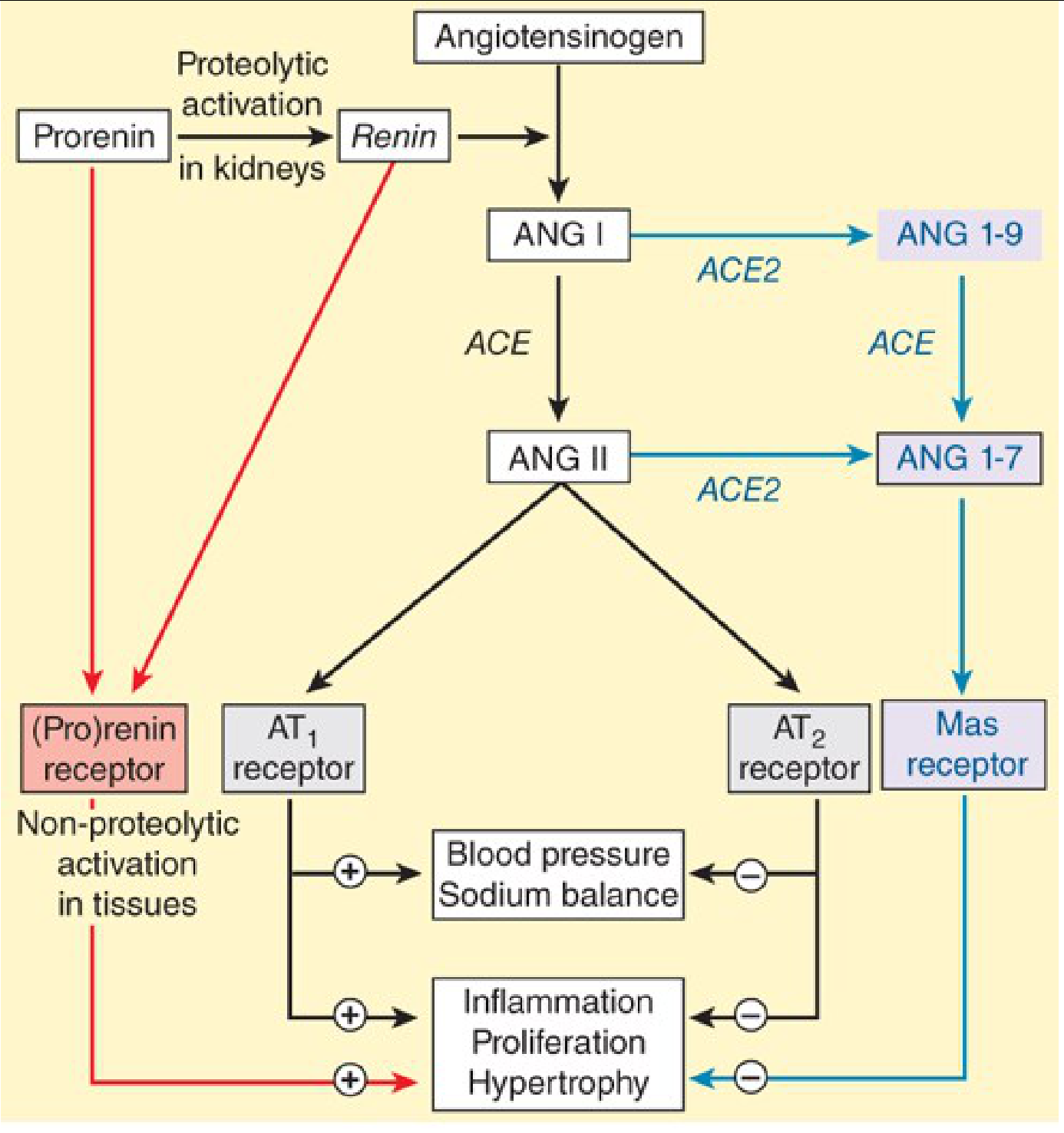
Full RAAS pathway including classical (black), (pro)renin receptor (red), and counter-regulatory Ang 1-7/Mas axis (blue) - Katzung's Basic and Clinical Pharmacology, 16th Ed.
Control of Renin Release
Renin release is the rate-limiting step of the RAAS - it is the primary determinant of overall system activity.
Stimuli for Renin Release
| Mechanism | Signal | Mediators |
|---|---|---|
| Macula densa | Decreased NaCl delivery to distal tubule (sensed via NKCC2) | PGE₂, NO (stimulate renin); adenosine (inhibits) |
| Renal baroreceptor | Decreased stretch of JG cells (low afferent arteriolar pressure) | Decreased Ca²⁺ influx into JG cells → increased renin |
| Sympathetic nervous system | Beta-1 adrenergic stimulation of JG cells (direct); also indirectly via baroreceptor and macula densa | Norepinephrine → ↑ cAMP → renin release |
| Low plasma Ang II | Negative feedback removed | Direct AT1R-mediated suppression absent |
Intracellular signaling in JG cells:
- cAMP is the primary driver: agents that raise cAMP increase renin (beta-agonists, PDE inhibitors, diuretics, vasodilators)
- Ca²⁺ is paradoxically inhibitory: unlike most secretory cells, increased intracellular Ca²⁺ in JG cells decreases renin release
- cGMP (via ANP, nitric oxide) inhibits renin via cGKII
Inhibitors of Renin Release
Ang II (negative feedback), nitric oxide, aldosterone, endothelin, adenosine, ANP, TNF-α, active Vitamin D, cGKII
Pharmacologic stimulators of renin release
Vasodilators (hydralazine, minoxidil, nitroprusside), beta-agonists, alpha-blockers, diuretics (including furosemide - which reduces NaCl delivery to macula densa), phosphodiesterase inhibitors
Angiotensin II Receptors
| Receptor | Location | Effects |
|---|---|---|
| AT1R (G-protein coupled) | Vascular smooth muscle, kidney (PT, efferent arteriole), adrenal cortex, heart, brain, liver | Vasoconstriction, Na⁺ retention, aldosterone release, cardiac hypertrophy, fibrosis, proinflammatory |
| AT2R | Fetal tissues, adrenal medulla, kidney | Vasodilation, natriuresis, anti-proliferative, anti-fibrotic (counter-regulatory) |
| Mas receptor | Brain, kidney, heart | Ang 1-7 binding → vasodilation, cardioprotection |
| (Pro)renin receptor (PRR) | Multiple tissues | Non-proteolytic activation of prorenin; downstream ERK1/2 MAP kinase signaling; pro-fibrotic effects |
Actions of Angiotensin II
Ang II acts primarily via AT1R:
Vascular
- Potent vasoconstrictor of systemic arterioles → ↑ total peripheral resistance (TPR) → ↑ blood pressure
- At low doses: preferential efferent arteriolar constriction → maintains GFR by increasing filtration fraction
- At high doses: both afferent and efferent constriction → ↓ RBF and GFR
- Chronic Ang II → glomerular hypertension → glomerular damage, proteinuria
Renal - Direct
- Stimulates Na⁺-H⁺ exchange (NHE3) in proximal tubule → increases Na⁺ and HCO₃⁻ reabsorption
- Upregulates Na⁺-K⁺-ATPase at basolateral membrane of proximal tubule
- Increases renovascular resistance, decreases medullary blood flow → mediates salt sensitivity
Renal - via Aldosterone
- Stimulates aldosterone synthesis and release from adrenal zona glomerulosa
- Aldosterone acts on principal cells of distal tubule and collecting duct → ↑ ENaC expression (Na⁺ reabsorption) and ↑ K⁺/H⁺ secretion
- Aldosterone's effects require gene transcription and new protein synthesis → slow response (hours to days)
Adrenal
- Stimulates aldosterone release (synergistic with hyperkalemia, which is an independent stimulus)
Central Nervous System
- Stimulates thirst and salt craving
- Stimulates ADH (AVP) secretion from posterior pituitary (via AT1R at the subfornical organ - a circumventricular organ lacking blood-brain barrier)
- These effects increase total body water → complement the Na⁺ retention effect
Cardiac / Vascular (Pathologic)
- Promotes cardiac hypertrophy (via AT1R, TGF-β pathway)
- Induces cardiac and vascular fibrosis, interstitial collagen remodeling
- Endothelial dysfunction, increased oxidative stress
- These maladaptive effects are mediated locally by tissue RAAS even when systemic Ang II levels are normal
Sympathoadrenal
- Augments catecholamine release and potentiates sympathetic activity
Aldosterone
- Produced by zona glomerulosa of adrenal cortex
- Stimulated by: Ang II, hyperkalemia (direct, synergistic with Ang II), ACTH (minor role)
- Acts on principal cells of cortical collecting duct:
- Increases apical ENaC expression → Na⁺ reabsorption
- Increases basolateral Na⁺-K⁺-ATPase
- Net effect: Na⁺ retention, K⁺ excretion, H⁺ excretion (metabolic alkalosis if excess)
- Local aldosterone in the heart: induces structural remodeling of interstitial collagen matrix → diastolic dysfunction, ventricular hypertrophy, fibrosis
Systemic vs. Local (Tissue) RAAS
A key modern concept is that there are local RAAS systems in the heart, kidney, brain, and vasculature, independent of the systemic (circulating) RAAS:
- Systemic RAAS is most prominent in acute decompensated states (acute HF, acute volume loss)
- Local/tissue RAAS dominates in chronic stable conditions (chronic HF, CKD, chronic hypertension)
- Local Ang II acts in a paracrine/autocrine manner via locally upregulated angiotensinogen and ACE
- This explains why RAAS inhibition remains effective in chronic HF even when circulating levels of Ang II are not markedly elevated
RAAS in Disease States
Heart Failure
- RAAS activation is proportional to severity of cardiac dysfunction and serves as a prognostic marker
- Initially adaptive: vasoconstriction maintains perfusion
- Chronically maladaptive: sodium retention, myocardial remodeling, fibrosis, progressive cardiac and renal dysfunction
- Local cardiac Ang II (via AT1R) → TGF-β mediated hypertrophy, fibrosis, reduced coronary flow
- Local aldosterone → interstitial fibrosis → systolic and diastolic dysfunction
Hypertension
- Ang II is central to BP dysregulation: acts via AT1R to cause systemic vasoconstriction, ↓ medullary blood flow, salt sensitivity
- Renal AT1R effects are more critical than extrarenal AT1R in sustained hypertension (cross-transplant experiments)
- Prorenin receptor (PRR) signaling also contributes via MAP kinase pathways
CKD
- Accumulation of endogenous organic acids impairs tubular secretion of RAAS-blocking drugs
- Chronic RAAS activation accelerates glomerulosclerosis and proteinuria
Pharmacologic Blockade of RAAS
| Drug Class | Site of Action | Notes |
|---|---|---|
| ACE inhibitors (enalapril, lisinopril, ramipril) | Block ACE → ↓ Ang II, ↓ aldosterone, ↑ bradykinin | Bradykinin accumulation → dry cough (10-15%), angioedema |
| ARBs (losartan, valsartan, candesartan) | Block AT1R directly | No bradykinin effect; no cough; used if ACE-I intolerant |
| Aldosterone antagonists / MRAs (spironolactone, eplerenone) | Block mineralocorticoid receptor | Potassium-sparing; eplerenone is more selective (fewer androgen side effects) |
| Direct renin inhibitors (aliskiren) | Block renin activity → ↓ Ang I and Ang II | No evidence of superiority over ACE-I or ARB alone; NOT combined with ACE-I/ARB |
| Beta-blockers | ↓ sympathetic stimulation of JG cells → ↓ renin release | Indirect RAAS suppression |
Clinical indications for RAAS blockade: Hypertension, heart failure (HFrEF), post-MI, diabetic nephropathy, proteinuric CKD, left ventricular hypertrophy, primary aldosteronism.
ACE-I/ARB combination: Dual RAAS blockade offers no additional renoprotection over monotherapy in most patients and increases risk of hypotension, hyperkalemia, and AKI - not routinely recommended.
Summary Diagram of the Full Cascade
Low BP / Low Na delivery / Sympathetic stimulation
↓
JG cells → RENIN released
↓
Angiotensinogen (liver) → ANGIOTENSIN I (decapeptide, inactive)
↓ [ACE - lung/endothelium]
ANGIOTENSIN II (octapeptide, highly active)
↙ ↓ ↓ ↘
AT1R AT1R AT1R AT1R
Vasoconstriction Proximal Adrenal Brain
↑TPR, ↑BP tubule Na⁺ Zona Thirst + ADH
reabsorption Glomerulosa ↑Water intake
↓
ALDOSTERONE
↓
Distal tubule/CD
↑ Na⁺ reabsorption
↓ K⁺/H⁺ excretion
↑ ECF volume → ↑BP
Counter-regulatory arm (ACE2):
ANG II → [ACE2] → ANG 1-7 → Mas receptor → Vasodilation, natriuresis
Sources: Katzung's Basic and Clinical Pharmacology 16th Ed.; Brenner and Rector's The Kidney; Costanzo Physiology 7th Ed.; Medical Physiology (Boron & Boulpaep); Comprehensive Clinical Nephrology 7th Ed.
Please give about loop diuretics and raas in five marks question type answers raas in a flow chart manner
Here are both topics in concise 5-mark answer format, with RAAS as a flowchart:
Loop Diuretics — 5 Mark Answer
Definition
Loop diuretics are the most potent class of diuretics, acting on the thick ascending limb (TAL) of the loop of Henle. Also called high-ceiling diuretics.
Drugs
- Furosemide (most common)
- Bumetanide
- Torsemide
- Ethacrynic acid (only non-sulfonamide - used in sulfa allergy)
Mechanism of Action
Site: Thick ascending limb (TAL) of loop of Henle
Action: Inhibit the Na⁺-K⁺-2Cl⁻ cotransporter (NKCC2) on the luminal membrane
- Blocks reabsorption of ~25% of filtered Na⁺
- Reduces lumen-positive potential → increases Mg²⁺ and Ca²⁺ excretion
- Disrupts medullary hypertonicity → impairs water reabsorption even in presence of ADH
- Increases PGE₂ synthesis (COX-2 induction) → contributes to diuresis and venodilation
- NSAIDs blunt this effect by inhibiting prostaglandin synthesis
Pharmacokinetics
| Drug | Oral Bioavailability | Duration |
|---|---|---|
| Furosemide | 12–112% (variable) | 2–3 hours |
| Torsemide | ~80–90% (consistent) | 4–6 hours |
| Bumetanide | ~80% | ~2 hours |
| Ethacrynic acid | ~100% | ~2 hours |
- All are highly protein-bound → reach tubular lumen via proximal tubule secretion (OAT1/OAT2)
- NSAIDs and probenecid compete for this transporter → reduced diuretic effect
Clinical Uses
- Acute pulmonary edema (IV furosemide - venodilation + diuresis)
- Chronic heart failure
- Nephrotic syndrome edema
- Hypercalcemia (with IV saline)
- Hypertension with GFR <30 mL/min or resistant hypertension
- Hyperkalemia, hyponatremia (life-threatening)
- CKD-associated edema
Adverse Effects
- Hypokalemia → cardiac arrhythmias (especially with digoxin)
- Hypomagnesemia
- Hypochloremic metabolic alkalosis
- Ototoxicity (tinnitus, deafness) - worst with ethacrynic acid and rapid IV use
- Volume depletion, hypotension
- Hyperuricemia, hyperglycemia
Drug Interactions
- Aminoglycosides / cisplatin → additive ototoxicity
- Digoxin → hypokalemia → digoxin toxicity
- NSAIDs / probenecid → reduced diuretic response
- Thiazides → synergistic diuresis (used in diuretic resistance)
RAAS — 5 Mark Answer (Flowchart Format)
Definition
The Renin-Angiotensin-Aldosterone System (RAAS) is the principal hormonal cascade regulating blood pressure, sodium balance, and extracellular fluid volume.
RAAS Flowchart
TRIGGER
─────────────────────────────────────────────────────
↓ Blood pressure / ↓ Na⁺ delivery to macula densa
/ ↑ Sympathetic activity (β1 stimulation)
│
▼
┌─────────────────┐
│ JG CELLS │ ← Juxtaglomerular cells
│ (Afferent │ in afferent arteriole
│ arteriole) │
└────────┬────────┘
│ releases
▼
RENIN
(aspartyl protease)
│
│ cleaves
▼
Angiotensinogen (liver) ──────────────────────────────────────────────►
│
▼
ANGIOTENSIN I
(decapeptide - INACTIVE)
│
│ ACE (lung endothelium)
│ + bradykinin inactivated here
▼
ANGIOTENSIN II
(octapeptide - ACTIVE)
│
┌────────────┼──────────────┬─────────────────┐
▼ ▼ ▼ ▼
AT1R on AT1R on AT1R in AT1R in
Arterioles Proximal Adrenal Brain
Tubule Cortex
│ │ │ │
▼ ▼ ▼ ▼
Vasoconstriction ↑ Na⁺-H⁺ ALDOSTERONE ↑ Thirst
↑ TPR → ↑ BP exchange released ↑ ADH release
↑ Na⁺ & HCO₃⁻ ↑ Water retention
reabsorption
│
▼
Distal tubule / Collecting duct
│
┌──────────┴──────────┐
▼ ▼
↑ ENaC activity ↑ Na⁺-K⁺-ATPase
↑ Na⁺ reabsorption ↑ K⁺ / H⁺ excretion
│
▼
↑ ECF volume → ↑ Blood volume
│
▼
┌──────────────────┐
│ ↑ BLOOD │
│ PRESSURE │
│ (RESTORED) │
└──────────────────┘
│
▼ (negative feedback)
↑ Ang II suppresses further renin release
↑ BP suppresses baroreceptor-mediated renin release
COUNTER-REGULATORY ARM (ACE2 pathway):
─────────────────────────────────────
Ang II ──[ACE2]──► Ang 1-7 ──► Mas receptor
│
▼
Vasodilation / Natriuresis
(opposes Ang II effects)
Key Points to Remember
| Component | Key Fact |
|---|---|
| Renin | Rate-limiting step; released from JG cells |
| Angiotensinogen | Substrate from liver; substrate concentration rarely rate-limiting |
| ACE | Lung endothelium; also inactivates bradykinin |
| Ang II | Acts via AT1R (harmful) and AT2R (protective/counter-regulatory) |
| Aldosterone | Slow response (hours-days); gene transcription required |
| ACE2 | Converts Ang II → Ang 1-7 (protective); entry point for SARS-CoV-2 |
Pharmacologic Blockade (for exam - 1 liner each)
| Drug Class | Example | Site Blocked |
|---|---|---|
| ACE inhibitors | Enalapril, Ramipril | Block ACE → ↓ Ang II, ↑ bradykinin (→ cough) |
| ARBs | Losartan, Valsartan | Block AT1R directly; no cough |
| MRAs | Spironolactone, Eplerenone | Block aldosterone receptor; K⁺-sparing |
| Direct renin inhibitor | Aliskiren | Blocks renin activity |
| Beta-blockers | Metoprolol | ↓ β1 stimulation → ↓ renin release |
Adverse Effects of RAAS Blockade (Exam Favourite)
- ACE inhibitors: Dry cough (bradykinin), hyperkalemia, angioedema, ↑ creatinine (especially with bilateral renal artery stenosis)
- ARBs: Hyperkalemia, ↑ creatinine; no cough
- Spironolactone: Gynecomastia, hyperkalemia
- Aliskiren: NOT combined with ACE-I or ARB (risk of hypotension, AKI, hyperkalemia)
This is a shared conversation. Sign in to Orris to start your own chat.