A 65 years old male on antipsychotic medications came with the C/o difficulty in walking, resting tremors and difficulty in swallowing a) What is the diagnosis. b)How will you treat this patient. c)Describe the mechanism of action, importance of combination therapy and adverse effects of Levodopa.
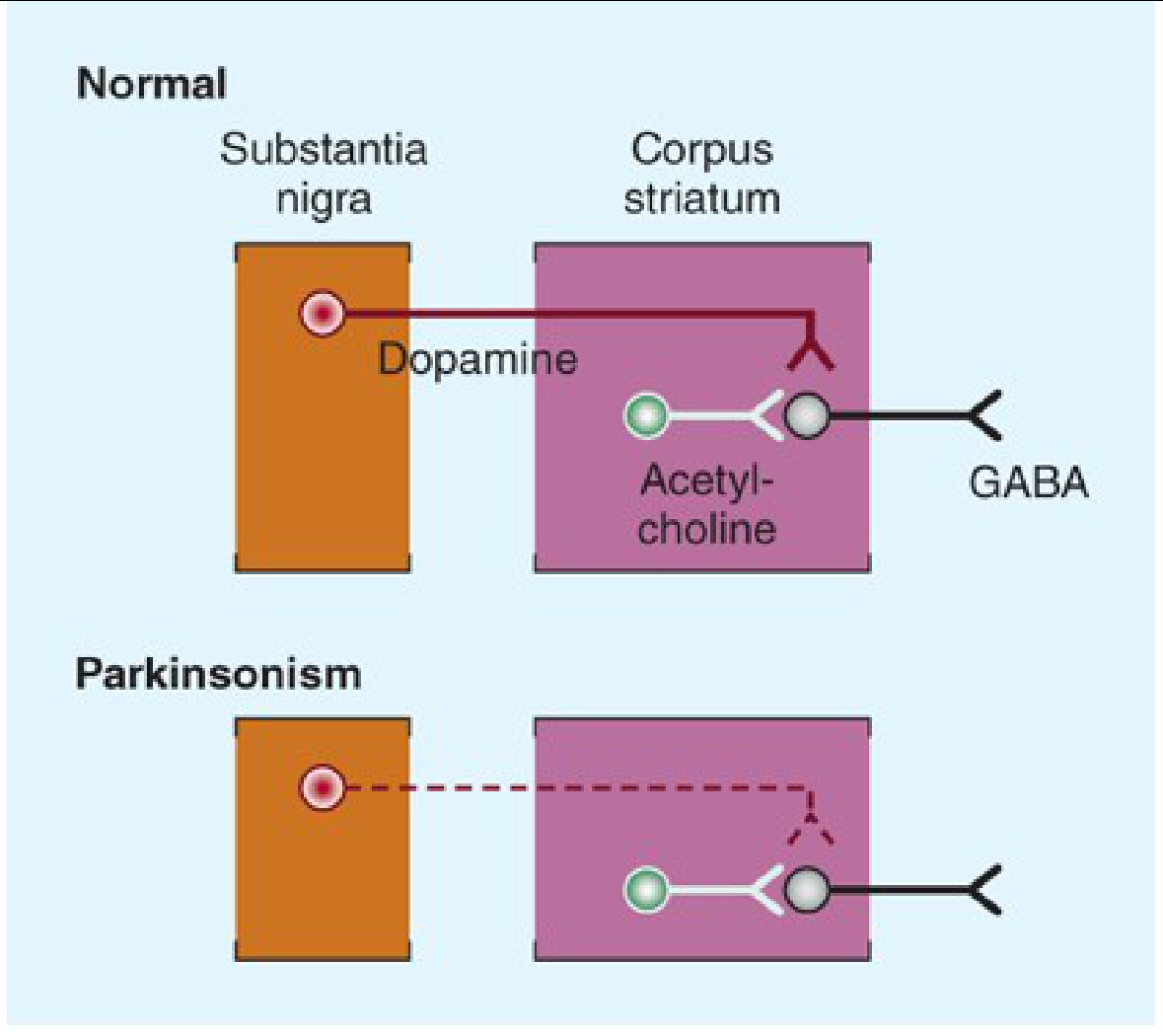
a) Diagnosis: Drug-Induced Parkinsonism (DIP)
- Resting tremors (pill-rolling type)
- Difficulty walking (bradykinesia, shuffling gait, rigidity)
- Dysphagia (difficulty swallowing - involvement of bulbar musculature)
b) Treatment
Step 1 - Reassess the antipsychotic
- If clinically possible, reduce the dose or switch to an atypical antipsychotic with lower D2-blocking potency (e.g., quetiapine, clozapine). These cause far less DIP.
- Note: Levodopa is NOT helpful if the neuroleptic drug is continued - it cannot overcome the D2 blockade and may aggravate the psychiatric disorder for which the antipsychotic was originally prescribed.
- If the antipsychotic is withdrawn, parkinsonian symptoms clear over several weeks to months. In elderly patients, this can take up to 3 months.
Step 2 - Symptomatic treatment for parkinsonism
| Drug | Mechanism | Notes |
|---|---|---|
| Anticholinergics (Benztropine/Cogentin, Trihexyphenidyl) | Restore dopamine-ACh balance | First-line for DIP; preferred treatment |
| Amantadine | Increases dopamine release, anticholinergic | Alternative option |
| Diphenhydramine (Benadryl) | Anticholinergic antihistamine | Useful, especially if IV/IM needed |
- Anticholinergics should be withdrawn after 4-6 weeks to assess whether tolerance to the parkinsonian effects has developed. About half of patients require continued treatment.
- Even after stopping the antipsychotic, continue anticholinergic until symptoms fully resolve.
c) Levodopa - Mechanism of Action, Combination Therapy, and Adverse Effects
Mechanism of Action
- Benefits primarily depend on stimulation of D2 receptors (postsynaptic striatal neurons), though D1 stimulation may also contribute to maximal effect
- Levodopa is particularly effective at relieving bradykinesia and related disabilities
Importance of Combination Therapy (Levodopa + Carbidopa)
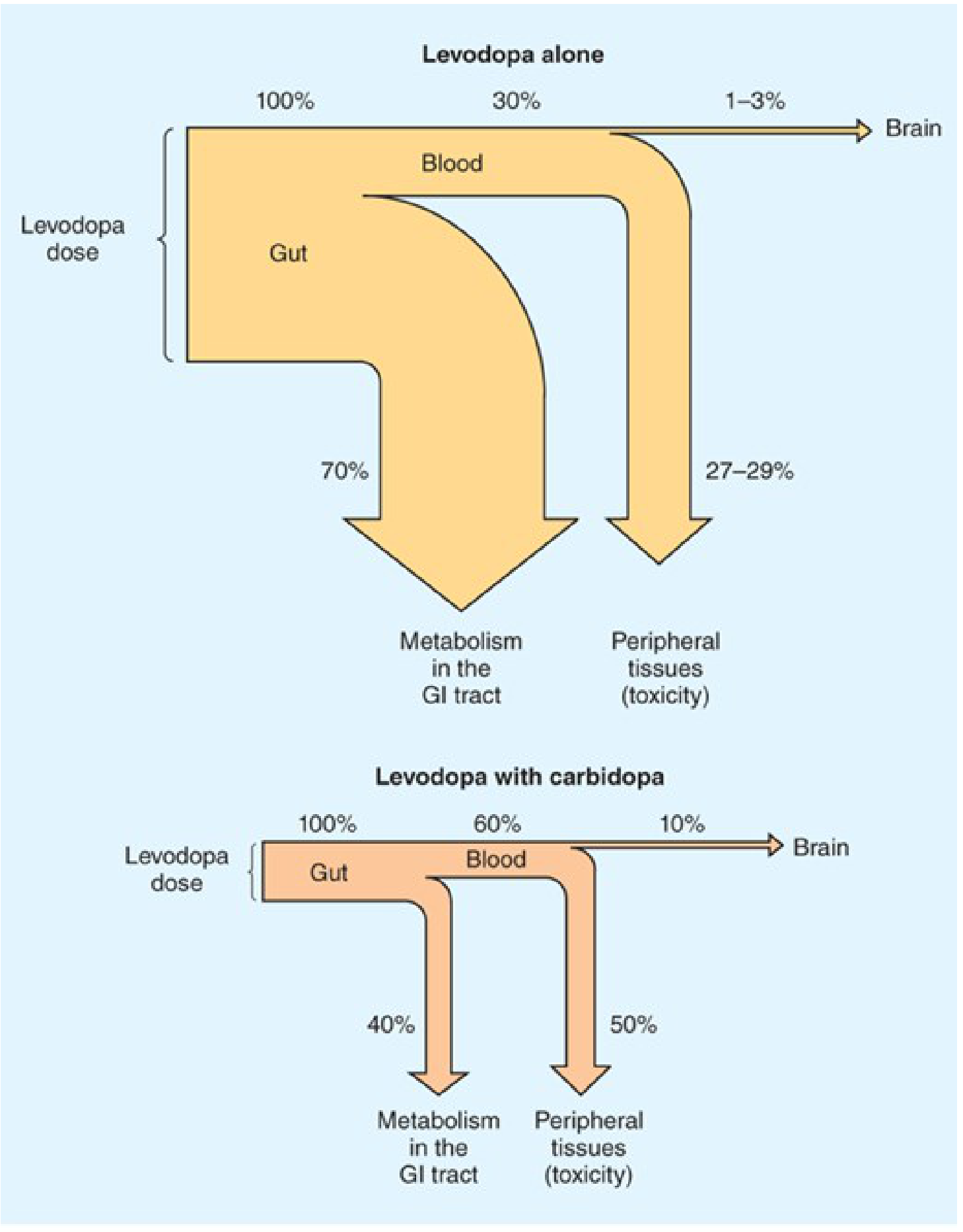
- Reduces daily levodopa requirement by ~75% - less drug needed for the same effect
- Plasma levels are higher and half-life longer - more drug available to reach the brain
- Brain delivery increases from ~1-3% to ~10% of the administered dose
- Dramatically reduces peripheral side effects - nausea/vomiting drops from ~80% to <20%; cardiac arrhythmias also reduced
- Allows much faster titration to therapeutic doses
Adverse Effects of Levodopa
- Anorexia, nausea, vomiting in ~80% when used alone (reduced to <20% with carbidopa)
- Due to stimulation of the chemoreceptor trigger zone (CTZ) - located outside the BBB
- Management: take with food, add extra carbidopa (Lodosyn 25 mg), use domperidone or ondansetron
- Avoid phenothiazine antiemetics - they worsen parkinsonism
- Cardiac arrhythmias (tachycardia, ventricular extrasystoles, rarely atrial fibrillation) - from peripheral catecholamine formation; reduced significantly with carbidopa co-administration
- Postural hypotension - common but often asymptomatic; tends to improve with continued treatment
- Hypertension - if combined with non-selective MAO inhibitors or sympathomimetics (dangerous combination)
- Depression, anxiety, agitation, insomnia, somnolence, confusion
- Delusions, hallucinations, nightmares, euphoria
- More common when levodopa is combined with carbidopa (more drug reaches the brain)
- Managed with atypical antipsychotics (quetiapine, clozapine) or pimavanserin (5-HT2A inverse agonist) for Parkinson psychosis
- Choreiform, athetoid, or dystonic movements - most common long-term complication
- Particularly affects the face, mouth, tongue, and limbs
- Related to peak plasma levodopa levels; managed by dose reduction or adding amantadine
- "Wearing-off" effect - symptom recurrence before next dose as plasma levodopa falls
- "On-off" phenomenon - unpredictable sudden shifts between mobility (on) and immobility (off), unrelated to dosing schedule
- Managed with: extended-release carbidopa/levodopa (Rytary), COMT inhibitors (entacapone, opicapone), MAO-B inhibitors (rasagiline), or deep brain stimulation
- Darkening of urine (levodopa metabolites - not harmful)
- Rarely, neuroleptic malignant-like syndrome on abrupt withdrawal
Summary Table
| Feature | Detail |
|---|---|
| Diagnosis | Drug-induced Parkinsonism (D2 blockade by antipsychotic) |
| First-line treatment | Reduce/switch antipsychotic + Anticholinergics (benztropine) |
| Why NOT levodopa for DIP? | Cannot overcome D2 blockade; may worsen psychiatric condition |
| Levodopa MOA | Precursor crosses BBB → converted to dopamine → D2 stimulation |
| Why add carbidopa? | Blocks peripheral decarboxylation → reduces dose by 75%, cuts GI/CV side effects |
| Key adverse effects | Nausea, arrhythmias, hypotension, hallucinations, dyskinesias, on-off fluctuations |
Neurohumoral transmission
Neurohumoral Transmission
1. Historical Background and Evidence
- The physiologically active compound and its biosynthetic enzymes are present at appropriate anatomical sites
- The compound can be recovered from perfusate of an innervated structure during nerve stimulation but not at rest
- The compound produces responses identical to nerve stimulation when administered exogenously
- Responses to nerve stimulation and the administered compound are modified identically by pharmacological antagonists
Note: Nitric oxide (NO) is an exception - it is a neurotransmitter in some postganglionic parasympathetic nerves but is synthesized on demand rather than stored and released by exocytosis.
2. Steps in Neurohumoral Transmission
Step 1 - Axonal Conduction
- At rest: interior is ~70 mV negative relative to exterior (resting membrane potential)
- Depolarization to threshold opens voltage-sensitive Na+ channels → rapid Na+ influx → depolarization with positive overshoot
- Followed by K+ efflux → repolarization
- Local circuit currents propagate the AP along the axon in a forward direction
- The depolarized region enters a brief refractory state to ensure unidirectional propagation
Step 2 - Junctional (Synaptic) Transmission
a) Storage and Release of Transmitter
- Non-peptide neurotransmitters (biogenic amines) are synthesized in axonal terminals and stored in synaptic vesicles
- Vesicular storage is driven by a vesicular proton pump (vesicular ATPase)
- Synaptic vesicle proteins include: synapsin, synaptophysin, synaptogyrin
- The exocytosis machinery involves SNARE proteins: synaptobrevin (vesicular membrane), SNAP-25 and syntaxin (plasma membrane), and synaptotagmin (Ca²+ sensor)
- Docking - Munc18 binds syntaxin, stabilizing the SNARE complex
- Priming I - Syntaxin assembles with SNAP-25, allowing synaptobrevin to bind
- Priming II - Complexin binds the SNARE complex; synaptotagmin binds Ca²+
- Fusion - Ca²+ entry triggers full membrane fusion and exocytosis
- Reset - NSF ATPase and SNAP adapters dissociate the SNARE complex
Botulinum toxin cleaves SNARE proteins (synaptobrevin) and blocks ACh release at the NMJ. Tetanus toxin acts similarly on inhibitory neurons in the CNS.
b) Combination with Postjunctional Receptor
- Released transmitter binds specific receptors on the postsynaptic membrane
- Ionotropic receptors (ligand-gated ion channels): rapid - opens within milliseconds
- Excitatory: nicotinic, glutamate, 5-HT3 → Na+ influx → EPSP (depolarization)
- Inhibitory: GABA, glycine → Cl- influx → IPSP (hyperpolarization)
- Metabotropic receptors (G protein-coupled): slower, via second messengers
- E.g., muscarinic receptors, α and β adrenergic receptors
- Signal through cAMP (adenylyl cyclase), IP3/DAG (phospholipase C), or modulation of K+/Ca²+ channels
c) Initiation of Response
- Summation of microscopic channel-opening events generates the excitatory postsynaptic potential (EPSP)
- Sufficient depolarization triggers a new action potential in the postjunctional cell
d) Termination of Transmitter Action
- Enzymatic hydrolysis (e.g., ACh by acetylcholinesterase)
- Reuptake into the nerve terminal (primary for catecholamines)
- Diffusion away from the synapse
e) Non-electrogenic (Trophic) Functions
- Neurotransmitters also control enzyme turnover, receptor density, and synaptic plasticity through trophic actions
3. Cholinergic Transmission
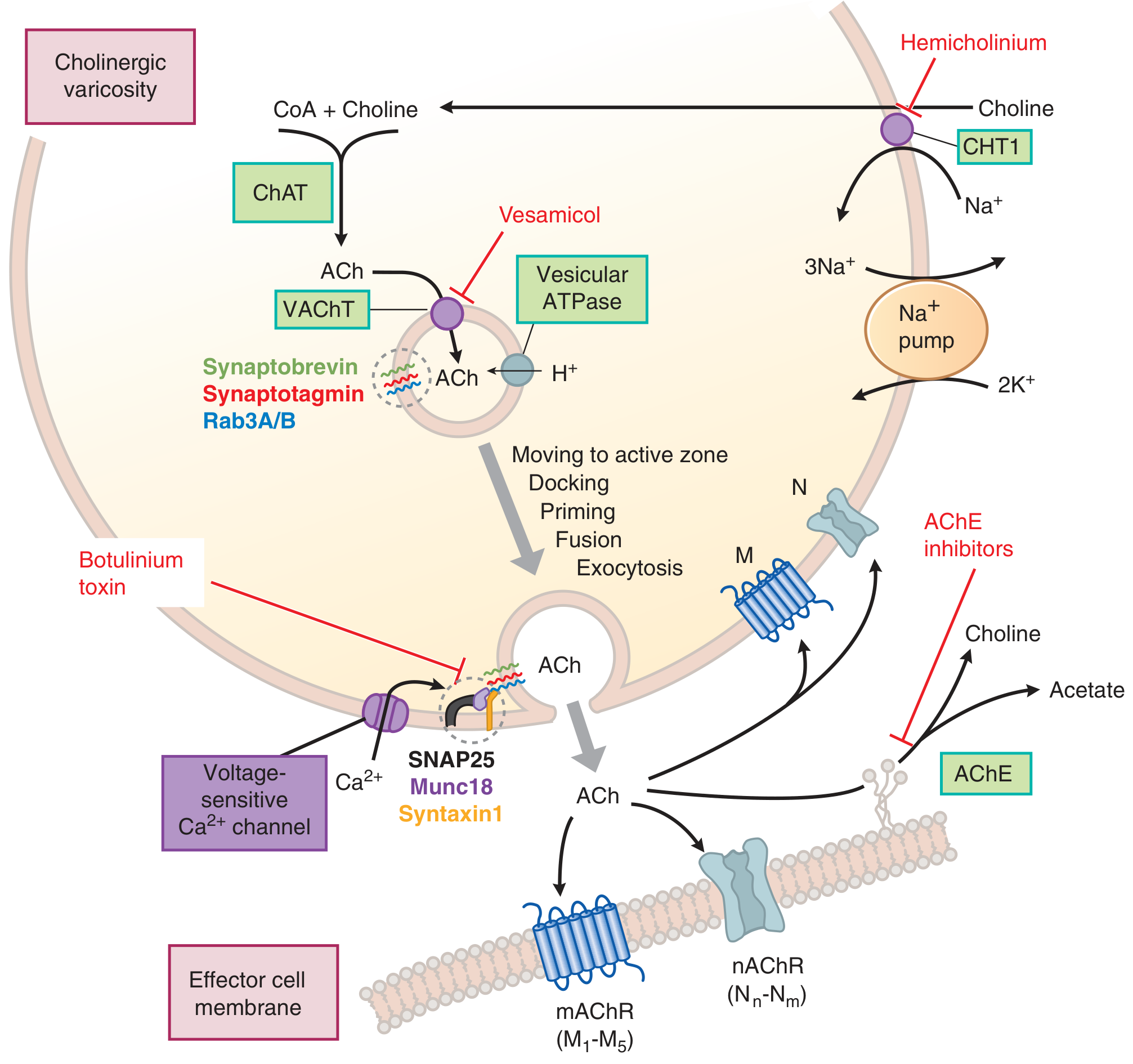
Synthesis of Acetylcholine (ACh)
- Choline acetyltransferase (ChAT) catalyzes: Choline + Acetyl CoA → ACh
- Rate-limiting step = uptake of choline via high-affinity choline transporter (CHT1) from the extracellular fluid (Na+-dependent)
- Blocked by: hemicholinium
- Choline is recycled after ACh hydrolysis
- ACh is packaged into vesicles by vesicular ACh transporter (VAChT)
- Blocked by: vesamicol
Release of ACh
- Action potential → Ca²+ entry via voltage-gated Ca²+ channels → SNARE-mediated exocytosis
- Two pools: readily releasable pool (near membrane, newly synthesized) and reserve pool (replenishes the first)
Degradation of ACh
- Acetylcholinesterase (AChE) hydrolyzes ACh → choline + acetate (within <1 millisecond at the NMJ)
- Choline is recycled back into the terminal
- Butyrylcholinesterase (pseudocholinesterase) - found in liver/plasma; physiologically hydrolyzes ingested plant esters
Cholinergic Receptors
| Receptor | Type | Location | Mechanism |
|---|---|---|---|
| Nicotinic (nAChR) | Ionotropic (ligand-gated Na+/K+ channel) | NMJ (Nm), Autonomic ganglia (Nn), CNS | EPSP/depolarization |
| Muscarinic M1,3,5 | Metabotropic (Gq) | Glands, smooth muscle, CNS | ↑ IP3/DAG → ↑ Ca²+ |
| Muscarinic M2,4 | Metabotropic (Gi) | Heart, presynaptic terminals | ↓ cAMP, ↑ K+ conductance |
4. Adrenergic (Catecholamine) Transmission
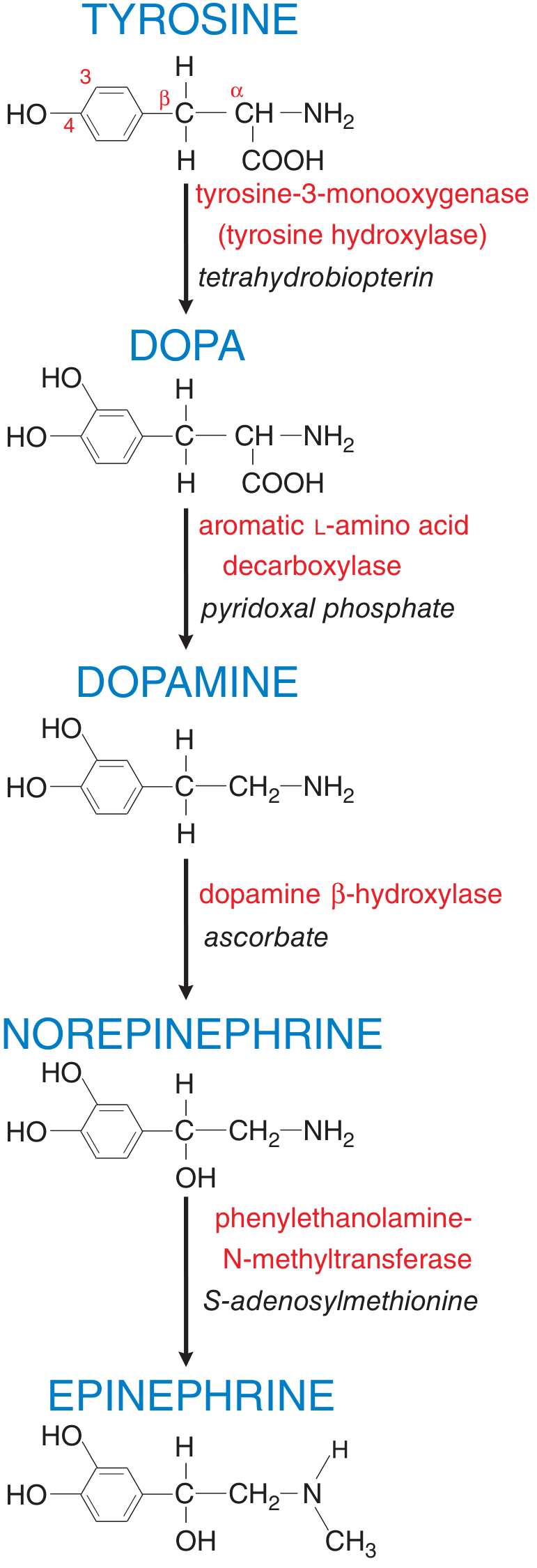
Synthesis of Norepinephrine (NE)
- Tyrosine → DOPA by tyrosine hydroxylase (TH) [rate-limiting step; cofactor: tetrahydrobiopterin]
- DOPA → Dopamine by aromatic L-amino acid decarboxylase (dopa decarboxylase) [cofactor: pyridoxal phosphate]
- Dopamine → Norepinephrine by dopamine β-hydroxylase (DβH) [cofactor: ascorbate] - inside storage vesicles
- Norepinephrine → Epinephrine by phenylethanolamine-N-methyltransferase (PNMT) [cofactor: S-adenosylmethionine] - only in adrenal medulla and a few brainstem neurons
Release of NE
- Action potential → Ca²+ entry → exocytosis of vesicular contents (NE, ATP, NPY, chromogranins, DβH)
- SNARE proteins (SNAP-25, syntaxin, synaptobrevin) mediate fusion
Termination of NE Action
| Transporter | Also called | Location | Affinity | Inhibitors |
|---|---|---|---|---|
| NET (SLC6A2) | Uptake 1 | Sympathetic nerves, adrenal medulla | High affinity for NE > EPI | Cocaine, TCAs (desipramine, imipramine) |
| OCT3/ENT (SLC22A3) | Uptake 2 | Nonneuronal cells (heart, liver) | Low affinity, prefers EPI > NE | Corticosterone, normetanephrine |
- MAO (monoamine oxidase) - intraneuronally; MAO-B selective for DA and phenylethylamines; MAO-A selective for 5-HT and NE
- COMT (catechol-O-methyltransferase) - extraneuronally
Adrenergic Receptors
| Receptor | G protein | Key Location | Effect |
|---|---|---|---|
| α1 | Gq | Smooth muscle, liver | Vasoconstriction, glycogenolysis |
| α2 | Gi | Presynaptic terminals, platelets | ↓ NE release (autoreceptor), platelet aggregation |
| β1 | Gs | Heart | ↑ Heart rate, ↑ contractility |
| β2 | Gs | Bronchi, blood vessels | Bronchodilation, vasodilation |
| β3 | Gs | Adipose tissue | Lipolysis |
5. Summary of Key Neurotransmitter Systems
| Feature | Cholinergic | Adrenergic |
|---|---|---|
| Neurotransmitter | Acetylcholine | Norepinephrine / Epinephrine |
| Precursor | Choline + Acetyl CoA | Tyrosine |
| Synthesis enzyme | Choline acetyltransferase (ChAT) | Tyrosine hydroxylase (rate-limiting) |
| Vesicular transporter | VAChT (blocked by vesamicol) | VMAT2 (blocked by reserpine) |
| Release trigger | Ca²+ entry via depolarization | Ca²+ entry via depolarization |
| Termination | Hydrolysis by AChE | Reuptake by NET; MAO/COMT metabolism |
| Receptors | Nicotinic (ionotropic), Muscarinic (metabotropic) | α1, α2, β1, β2, β3 (all metabotropic/GPCR) |
| Blocked by (release) | Botulinum toxin | - |
| Blocked by (synthesis) | Hemicholinium (choline uptake) | α-methyltyrosine (TH inhibitor) |
25 years male RTA victim presented with rapid pulse, cold clammy cyanotic skin, tachypnea and landed in emergency ward in unconscious state what is the diagnosis? Classify the disease write in detail about the pathogenesis, morphology evolution of the disease.
Diagnosis: Hypovolemic Shock (Hemorrhagic Shock)
- Rapid (tachycardic) pulse - compensatory response to falling cardiac output
- Cold, clammy, cyanotic skin - cutaneous vasoconstriction shunting blood to vital organs
- Tachypnea - compensatory hyperventilation to improve oxygenation and blow off CO2
- Unconsciousness - end-organ (brain) hypoperfusion
Classification of Shock
| Type | Clinical Example | Principal Mechanism |
|---|---|---|
| Hypovolemic | Hemorrhage, burns, vomiting, diarrhea, trauma | Inadequate blood or plasma volume → low cardiac output |
| Cardiogenic | Myocardial infarction, arrhythmia, cardiac tamponade, pulmonary embolism | Myocardial pump failure → low cardiac output |
| Septic | Gram-positive/negative sepsis, fungi | Peripheral vasodilation, vascular leak, DIC, cytokine cascade |
| Neurogenic | Spinal cord injury, general anesthesia | Loss of vascular tone → acute vasodilation → hypotension |
| Anaphylactic | IgE-mediated hypersensitivity | Systemic vasodilation + increased vascular permeability |
Pathogenesis of Shock
Core Mechanism
Massive hemorrhage → ↓ circulating blood volume → ↓ venous return → ↓ cardiac output → ↓ tissue perfusion → cellular hypoxia → organ dysfunction → death (if uncorrected)
Compensatory (Neurohumoral) Responses Activated
- Baroreceptor reflexes - carotid/aortic baroreceptors detect falling BP → reflex sympathetic activation
- Catecholamine release (epinephrine from adrenal medulla, NE from sympathetics) → tachycardia, peripheral vasoconstriction
- Renin-Angiotensin-Aldosterone System (RAAS) activation → angiotensin II causes vasoconstriction; aldosterone promotes renal Na+ and water retention
- ADH (vasopressin) release from posterior pituitary → water reabsorption by kidney
- Generalized sympathetic stimulation → redistributes blood away from skin, gut, and kidneys toward heart and brain
- Tachycardia
- Peripheral vasoconstriction (cold, clammy, pale/cyanotic skin)
- Oliguria (urine output falls)
- Relative preservation of cardiac and cerebral perfusion initially
Evolution (Stages) of Shock
Stage 1 - Nonprogressive (Compensated) Stage
- Reflex compensatory mechanisms are fully activated
- Vital organ (heart, brain) perfusion is maintained
- Clinically: tachycardia, tachypnea, cool clammy skin, mild anxiety
- Cellular injury is reversible at this stage
- Blood is shunted away from skin/gut to heart and brain
- Cutaneous vasoconstriction accounts for the cold, clammy, cyanotic appearance
Stage 2 - Progressive Stage
- Underlying cause is not corrected → widespread tissue hypoxia develops
- Aerobic respiration fails → anaerobic glycolysis → lactic acid production → metabolic lactic acidosis
- Lactic acidosis lowers tissue pH → vasomotor response is blunted
- Arterioles dilate → blood pools in the microcirculation
- Peripheral pooling worsens cardiac output (vicious cycle)
- Endothelial anoxic injury → risk of Disseminated Intravascular Coagulation (DIC)
- Vital organs (heart, brain, kidney) begin to fail
- Clinically: worsening hypotension, deepening coma, oliguria/anuria, worsening lactic acidosis
Stage 3 - Irreversible Stage
- Severe, sustained tissue injury → widespread cell death
- Lysosomal enzyme leakage from necrotic cells further worsens the shock state (autolytic injury)
- Myocardial contractile function deteriorates (partly from excess NO synthesis reducing cardiac muscle tone)
- Ischemic bowel allows gut bacteria to enter the circulation → superimposed bacteremic shock (septic shock on top of hemorrhagic shock)
- Acute tubular necrosis (ATN) of kidneys from sustained ischemia → acute renal failure
- Despite heroic therapeutic efforts, death is inevitable once this stage is reached
- The terminal pathway is multi-organ failure (MOF)
Morphology of Shock
1. Brain
- Ischemic encephalopathy - neuronal necrosis in the most vulnerable areas: hippocampus (Sommer's sector), cerebellar Purkinje cells, and neocortical neurons
- Watershed (border-zone) infarcts in areas between the end-territories of major cerebral arteries (ACA-MCA and MCA-PCA junction zones)
- Neurons are the most sensitive cells to hypoxia - irreversible damage after just 3-5 minutes of complete anoxia
2. Heart
- Subendocardial hemorrhage and necrosis - the subendocardial myocardium is the zone most vulnerable to ischemia (farthest from coronary supply, highest oxygen demand)
- Focal areas of myocardial necrosis (contraction band necrosis) - seen in hemorrhagic shock
- Reduced myocardial contractility with worsening shock (partly mediated by TNF, IL-1, and NO in later stages)
3. Kidneys
- Acute Tubular Necrosis (ATN) - the most characteristic morphological finding
- Patchy ischemic necrosis of tubular epithelium, especially the proximal tubule and thick ascending limb of Henle
- Tubular casts (pigmented) in the distal tubules and collecting ducts
- Interstitial edema
- Intact tubular basement membrane (important - allows regeneration if patient survives)
- Clinically: oliguria, rising BUN and creatinine, electrolyte disturbances
- Prognosis: tubular cells can regenerate - reversible if perfusion is restored
4. Lungs
- Lungs are relatively resistant to hypoxic injury in pure hemorrhagic shock
- However, when shock is complicated by trauma or sepsis: Diffuse Alveolar Damage (DAD) develops
- Interstitial and alveolar edema
- Hyaline membrane formation (protein-rich exudate)
- Type II pneumocyte hyperplasia
- This is the pathological substrate of "Shock Lung" = Acute Respiratory Distress Syndrome (ARDS)
- Clinically: hypoxia, bilateral infiltrates on CXR, respiratory failure
5. Adrenal Glands
- Cortical cell lipid depletion - a non-specific stress response reflecting increased use of stored cholesterol/lipids for cortisol synthesis
- In severe septic shock complicated by DIC: adrenal hemorrhage → Waterhouse-Friderichsen syndrome → adrenal insufficiency
- Loss of cortisol in late stages contributes to hemodynamic collapse
6. Gastrointestinal Tract
- Mucosal ischemia → stress ulcers (Curling's ulcers in the stomach/duodenum)
- Mucosal barrier breakdown → bacterial translocation (gut bacteria enter the bloodstream)
- This is a key mechanism for converting hypovolemic shock into septic shock in the later stages
- Can also develop hemorrhagic gastroenteritis and ischemic colitis
7. Liver
- Centrilobular necrosis (zone 3 of hepatic acinus - most distal from hepatic artery supply, most sensitive to hypoxia)
- "Nutmeg liver" appearance grossly (congested, red-yellow mottled)
- Rising liver enzymes (transaminases) → jaundice in severe/prolonged shock
Summary Flowchart
RTA → Hemorrhage
↓
↓ Circulating Blood Volume
↓
↓ Cardiac Output
↓
↓ Tissue Perfusion
↓
┌──────────────────────────────────────────┐
│ COMPENSATORY STAGE │
│ ↑ HR, ↑ Catecholamines, ↑ RAAS, ↑ ADH │
│ Peripheral vasoconstriction │
│ Cold clammy cyanotic skin │
└──────────────────────────────────────────┘
↓ (if not corrected)
┌──────────────────────────────────────────┐
│ PROGRESSIVE STAGE │
│ Anaerobic glycolysis → Lactic acidosis │
│ Arteriolar dilation → Microvascular │
│ pooling → DIC risk │
│ Vital organ dysfunction begins │
└──────────────────────────────────────────┘
↓ (if not corrected)
┌──────────────────────────────────────────┐
│ IRREVERSIBLE STAGE │
│ Lysosomal rupture, ↓ myocardial │
│ contractility, gut translocation, │
│ ATN, ARDS, MOF → DEATH │
└──────────────────────────────────────────┘
Clinical Note
Prognosis: More than 90% of young, otherwise healthy patients with hypovolemic shock survive with appropriate management (aggressive IV fluid resuscitation, blood transfusion, surgical hemorrhage control). This compares favorably with cardiogenic or septic shock, which carry substantially worse outcomes even with state-of-the-art care. The key is rapid intervention before the irreversible stage is reached.
20 years male following antibiotic intake had total leukocyte count of 1200 cells / mm3 and absolute neutrophil count of 120 cells. What is your diagnosis? What is the pathogenesis of this condition?
Diagnosis
Primary Diagnosis: Drug-Induced Agranulocytosis
| Parameter | Patient's Value | Normal Range | Interpretation |
|---|---|---|---|
| Total Leukocyte Count (TLC) | 1200/mm³ | 4000-11,000/mm³ | Severe leukopenia |
| Absolute Neutrophil Count (ANC) | 120/mm³ | 1500-8000/mm³ | Agranulocytosis (<500/mm³) |
- Neutropenia = ANC < 1500/mm³
- Severe neutropenia = ANC < 500/mm³
- Agranulocytosis = ANC < 100-500/mm³ (marked reduction, near absence of neutrophils)
Classification of Neutropenia/Agranulocytosis
Broad Mechanism-Based Classification
AGRANULOCYTOSIS
├── A. Decreased Production (Inadequate/Ineffective Granulopoiesis)
│ ├── 1. Suppression of Hematopoietic Stem Cells (HSCs)
│ │ ├── Aplastic anemia
│ │ ├── Marrow infiltration (leukemia, granulomas, tumors)
│ │ └── Myelotoxic chemotherapy (affects all cell lines)
│ ├── 2. Suppression of Committed Granulocytic Precursors
│ │ └── Drug toxicity (selective - other lineages spared)
│ ├── 3. Ineffective Hematopoiesis
│ │ ├── Megaloblastic anemia (B12/folate deficiency)
│ │ └── Myelodysplastic neoplasms
│ └── 4. Congenital
│ └── Kostmann syndrome (severe congenital neutropenia)
│
└── B. Increased Destruction/Sequestration
├── 1. Immune-mediated neutrophil destruction (drug-induced or idiopathic)
├── 2. Splenomegaly (splenic sequestration)
├── 3. Overwhelming infection (bacterial, fungal, rickettsial)
└── 4. LGL leukemia (CD8+ cytotoxic T cell suppression of myelopoiesis)
Drug-Based Classification of Causes
| Mechanism | Drug Class | Examples |
|---|---|---|
| Predictable, dose-related myelosuppression | Antineoplastics | Alkylating agents, antimetabolites |
| Idiosyncratic - direct toxic to precursors | Antipsychotics | Chlorpromazine, clozapine, phenothiazines |
| Idiosyncratic - immune-mediated | Antibiotics | Penicillins, sulfonamides, chloramphenicol |
| - | Antithyroidal | Methimazole, propylthiouracil, carbimazole |
| - | Anticonvulsants | Valproate, carbamazepine |
| - | Anti-inflammatory | Sulfasalazine |
| - | Antiarrhythmic | Procainamide |
Pathogenesis
Mechanism 1: Direct Toxic/Myelosuppressive Effect
Drug enters bone marrow
↓
Direct toxic effect on granulocytic precursors
(myeloblasts, promyelocytes, myelocytes)
↓
Selective destruction of granulocyte precursors
(erythroid and megakaryocytic lineages spared)
↓
Maturation arrest at promyelocyte/myelocyte stage
↓
↓↓ Release of mature neutrophils into blood
↓
Agranulocytosis
Mechanism 2: Immune-Mediated (Hapten) Mechanism
STEP 1: SENSITIZATION PHASE
Drug (hapten) binds to neutrophil surface proteins
→ Forms drug-protein complex (neoantigen)
→ Presented to immune system as foreign antigen
→ Antibody production against drug-neutrophil complex
(IgG/IgM anti-neutrophil antibodies generated)
STEP 2: SUBSEQUENT EXPOSURE
Re-exposure to the same drug
↓
Drug binds to neutrophil surface again
↓
Pre-formed antibodies (IgG) attach to drug-neutrophil complex
↓
Two pathways of destruction:
├─ COMPLEMENT ACTIVATION (IgM/IgG):
│ Complement fixed on neutrophil surface
│ → Membrane Attack Complex (MAC)
│ → Direct neutrophil lysis in circulation
│
└─ OPSONIZATION + PHAGOCYTOSIS (IgG):
Fc receptors on macrophages/monocytes
recognize IgG-coated neutrophils
→ Phagocytosis in spleen and liver
→ Peripheral neutrophil destruction
↓
Rapid fall in circulating neutrophils
↓
AGRANULOCYTOSIS
Additional Immune Mechanism: Autoimmune Neutropenia
- Antibodies directed against neutrophil surface antigens
- Idiopathic or associated with SLE, rheumatoid arthritis (Felty syndrome)
- Neutrophils opsonized → destroyed in spleen
Morphology
Bone Marrow Changes
| Type of Mechanism | Bone Marrow Appearance |
|---|---|
| Immune-mediated peripheral destruction | Hypercellular - compensatory increase in granulocytic precursors (the marrow tries to compensate for peripheral loss) |
| Direct toxic suppression of precursors | Hypocellular (specifically reduced granulocytic series; erythroid and megakaryocytes preserved) |
| Myelotoxic chemotherapy | Hypocellular all lineages reduced |
Peripheral Blood
- Near-absent neutrophils (ANC <500)
- Other cell lines (RBCs, platelets, lymphocytes) relatively preserved in pure drug-induced cases
Sites of Infection (Consequences of Agranulocytosis)
- Oral cavity most characteristic: Necrotizing ulcerative lesions of the gingiva, floor of mouth, buccal mucosa, and pharynx
- Deep, undermined ulcers covered by gray-to-green-black necrotic membranes
- Masses of bacteria/fungi visible histologically with minimal leukocytic inflammatory response (no neutrophils available)
- Lungs, urinary tract, kidneys: Severe invasive bacterial or fungal infections
- Deep fungal infections: Candida and Aspergillus (characteristically opportunistic)
- All sites share the hallmark: massive microbial growth with absent neutrophilic infiltrate
Clinical Features and Danger Threshold
| ANC | Risk Level | Clinical Implication |
|---|---|---|
| 1000-1500/mm³ | Mild | Increased susceptibility to infection |
| 500-1000/mm³ | Moderate | Significant infection risk |
| <500/mm³ | Severe | Risk rises sharply - infections become life-threatening |
| <100/mm³ | Agranulocytosis | Overwhelming infections; can cause death within hours to days |
- Malaise, chills, high-grade fever (often the first sign)
- Oropharyngeal ulcers/mucositis
- Weakness and fatigue
- If untreated: rapidly progressive sepsis → death
Management Principles
- Stop the offending drug immediately (causative antibiotic)
- Broad-spectrum antibiotics at first sign of infection (do not wait for culture results - infections are fulminant)
- G-CSF (Granulocyte Colony-Stimulating Factor / Filgrastim) - stimulates granulocyte production from marrow precursors, accelerates recovery
- Reverse isolation (protective environment to minimize infection exposure)
- Bone marrow biopsy to assess cellularity and mechanism
- Antifungal prophylaxis (fluconazole/voriconazole) given risk of Candida/Aspergillus
Prognosis: With early diagnosis, cessation of the offending drug, and G-CSF therapy, most patients recover. Mortality is high if diagnosis is delayed and fulminant sepsis develops.