Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
acute respiratory distress syndrome ARDS pathogenesis mechanism diagram
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have rich, comprehensive content from three major textbooks. Let me compile the full explanation.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
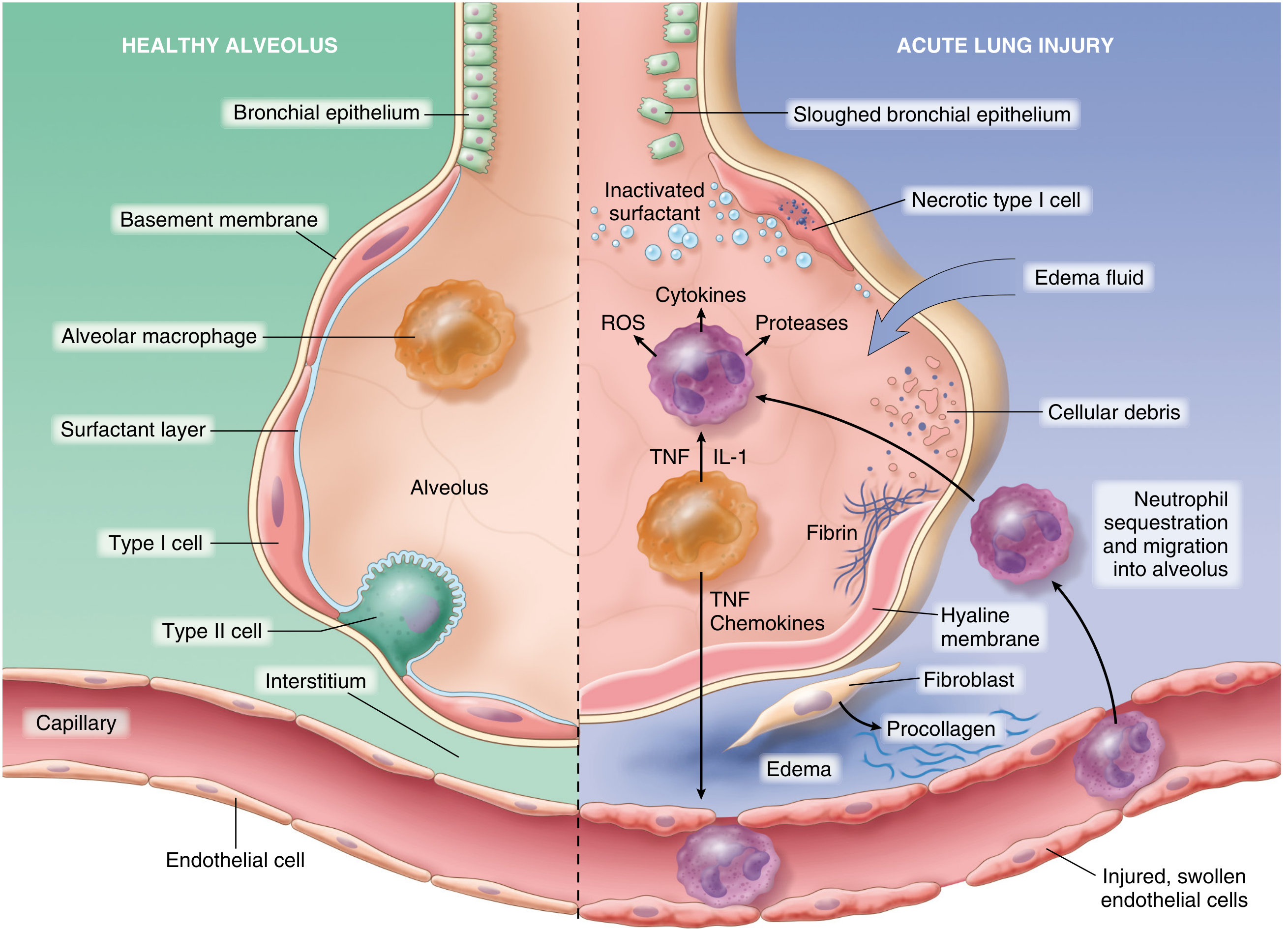
The normal alveolus (left) versus the injured alveolus in ARDS (right), showing the key cellular events — Robbins Pathologic Basis of Disease
Definition and Core Concept
ARDS is noncardiogenic pulmonary edema accompanied by severe lung inflammation, hypoxemia, and decreased lung compliance leading to respiratory failure. Unlike cardiogenic pulmonary edema driven by elevated hydrostatic pressure, ARDS edema is exudative — the alveolar-capillary barrier becomes hyperpermeable and leaks protein-rich fluid into the airspaces. — Murray & Nadel's Textbook of Respiratory Medicine
Triggers (Initiating Insults)
ARDS arises from both direct (pulmonary) and indirect (extrapulmonary) insults. Over 50% of cases are attributable to four conditions:
| Direct | Indirect |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis |
| Gastric aspiration | Severe trauma / head injury |
| Pulmonary contusion | Pancreatitis |
| Near-drowning | Transfusion-related lung injury (TRALI) |
| Inhalation injury | DIC, burns, drug overdose |
— Robbins Pathologic Basis of Disease
Pathogenesis: Step-by-Step
1. Initial Alveolar-Capillary Injury
The cascade begins with injury to type I alveolar pneumocytes (gas exchange cells) and pulmonary capillary endothelium, triggered by the insult above. Resident alveolar macrophages sense this damage and respond by releasing early cytokines — particularly TNF-α and IL-1 — that activate the neighboring endothelium and recruit circulating leukocytes. In systemic insults (sepsis, trauma), circulating inflammatory mediators may directly activate pulmonary endothelium without prior pneumocyte injury. — Robbins
2. Neutrophil Recruitment and Activation
Endothelial activation upregulates adhesion molecules (ICAM-1, E-selectin, P-selectin) on the capillary surface. Circulating neutrophils adhere, marginate, and extravasate into the interstitium and alveolar space. Once there, neutrophils:
- Release proteases (elastase, matrix metalloproteinases) that digest the basement membrane
- Generate reactive oxygen species (ROS) that damage cellular membranes and DNA
- Secrete pro-inflammatory cytokines (IL-8, IL-6, TNF-α) amplifying the inflammatory loop
- Form neutrophil extracellular traps (NETs) that directly damage lung tissue
This creates a self-amplifying feedback loop: inflammation → endothelial damage → more neutrophil recruitment. — Murray & Nadel's, Robbins
3. Breakdown of the Alveolar-Capillary Barrier
The combined injury from neutrophil proteases, ROS, and cytokines disrupts both:
- Pulmonary capillary endothelium → loss of tight junctions → interstitial edema
- Type I pneumocytes (which cover ~95% of alveolar surface) → necrosis → denuded basement membrane
The result is flooding of alveoli with protein-rich exudate containing fibrin, serum proteins, red cells, and cellular debris. This is the hallmark of diffuse alveolar damage (DAD). — Murray & Nadel's
4. Surfactant Dysfunction
Type II pneumocytes produce surfactant, which normally reduces alveolar surface tension and prevents collapse. In ARDS:
- Type II cells are injured or overwhelmed
- Surfactant proteins are diluted and inactivated by the flooding edema fluid
- Phospholipase A₂ (released during pancreatitis-associated ARDS) enzymatically degrades surfactant
Loss of surfactant leads to alveolar collapse (atelectasis) and further worsens hypoxemia. — Murray & Nadel's, Murray & Nadel's (pancreatitis chapter)
5. Hyaline Membrane Formation
Over the first several days (exudative phase, days 1–7), the protein-rich intraalveolar fluid condenses with fibrin and necrotic cellular debris to form hyaline membranes — eosinophilic, glassy deposits lining the alveolar walls. This is the pathognomonic finding of DAD on histology. Hyaline membranes impair gas diffusion and further reduce lung compliance. — Murray & Nadel's, Robbins
6. Coagulation Dysregulation
Inflammatory activation simultaneously triggers intravascular coagulation pathways:
- Endothelial activation induces expression of procoagulant proteins (tissue factor) and reduces expression of anticoagulant thrombomodulin
- Intravascular fibrin deposition within pulmonary capillaries contributes to pulmonary hypertension by obstructing the microcirculation
- Platelet-activating factor (PAF) and platelet-neutrophil complexes (via P-selectin/PSGL-1) amplify both coagulation and ROS release
7. Impaired Alveolar Fluid Clearance
Normally, alveolar epithelial Na⁺ channels (ENaC) drive Na⁺ reabsorption, creating an osmotic gradient that clears edema fluid, with Na⁺/K⁺-ATPases maintaining the gradient. In ARDS:
- Hypoxia impairs ENaC expression and Na⁺/K⁺-ATPase activity
- Nitric oxide (elevated in sepsis and shock) disrupts β-adrenergic-mediated upregulation of alveolar fluid clearance
- This impairs the lung's ability to resolve edema, perpetuating flooding
8. Angiopoietin Imbalance
Angiopoietin-2 (Ang2), expressed by activated endothelial cells, competes with the barrier-stabilizing Angiopoietin-1 (Ang1) at the Tie2 receptor. Elevated Ang2 levels are found in sepsis and ARDS, and exogenous Ang2 induces pulmonary vascular leak in animal models. Ang2 is considered both a biomarker and a pathogenic mediator of vascular barrier dysfunction in ARDS. — Murray & Nadel's
Phases of ARDS (Histopathologic Progression)
| Phase | Timing | Key Features |
|---|---|---|
| Exudative | Days 1–7 | Hyaline membranes, neutrophilic infiltration, protein-rich edema, DAD |
| Proliferative | Days 7–21 | Hyaline membrane reorganization, type II pneumocyte hyperplasia, early fibrosis |
| Fibrotic | >2–3 weeks | Collagen deposition, obliteration of pulmonary capillaries, fibrosis in ~30% |
Type II alveolar cell hyperplasia during the proliferative phase attempts to resurface denuded alveoli and produce new surfactant. If inflammation abates, macrophages clear debris and release TGF-β and PDGF, stimulating fibroblast proliferation and collagen deposition. — Murray & Nadel's, Robbins
Physiologic Consequences
| Derangement | Mechanism |
|---|---|
| Refractory hypoxemia | Alveolar flooding + atelectasis → intrapulmonary shunt (perfusion without ventilation) |
| Reduced lung compliance | Alveolar edema + hyaline membranes + alveolar collapse |
| Pulmonary hypertension | Hypoxic vasoconstriction + microvascular fibrin + compression by positive-pressure ventilation |
| Increased dead space | Microvascular obliteration → ventilation without perfusion |
| Ventilator-induced lung injury (VILI) | Overdistension of remaining aerated alveoli generates barotrauma/volutrauma, further amplifying cytokine release |
Ventilator-Induced Lung Injury (VILI)
Because ARDS lungs are heterogeneous — with consolidated dependent regions and relatively normal non-dependent regions — positive-pressure ventilation with standard tidal volumes overdistends the "baby lung" of aerated units. This generates biotrauma (cytokine release from mechanically stressed alveolar cells) in addition to barotrauma, which can propagate systemic organ failure. This is the rationale for lung-protective ventilation (low tidal volume 6 mL/kg predicted body weight, plateau pressure <30 cmH₂O). — Murray & Nadel's
Summary Pathway
Insult (sepsis, pneumonia, aspiration, trauma)
↓
Pneumocyte/Endothelial Injury
↓
Macrophage Activation → TNF-α, IL-1, IL-8
↓
Neutrophil Recruitment → Proteases, ROS, NETs
↓
Alveolar-Capillary Barrier Disruption
↓
Protein-rich Edema + Surfactant Loss + Hyaline Membranes (DAD)
↓
Coagulation Activation + Ang2↑ → Microvascular Thrombosis
↓
Impaired Na⁺/fluid clearance → Persistent Edema
↓
Hypoxemia (Shunt) + Reduced Compliance + Pulmonary Hypertension
↓
Fibroproliferation (TGF-β, PDGF) → Fibrosis (subset)
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine — Chapter 134 (ARDS) and Chapter 32
- Robbins & Cotran Pathologic Basis of Disease — Chapter 15 (ALI/ARDS)
- Murray & Nadel's — Chapter 22 (Diffuse Alveolar Damage histopathology)
This is a shared conversation. Sign in to Orris to start your own chat.