Umn vs lmn lesions
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Excellent - I now have all the data needed for a thorough, well-sourced answer. Let me compile it.
UMN vs LMN Lesions
Overview
The motor system has two levels of neurons in series:
- Upper Motor Neuron (UMN): Cell bodies in the motor cortex (precentral gyrus); axons travel via the corticospinal and corticobulbar tracts to synapse on lower motor neurons. Also includes brainstem neurons that control spinal motor neurons.
- Lower Motor Neuron (LMN): Cell bodies in the anterior horn of the spinal cord (or cranial nerve motor nuclei for the head); axons travel via peripheral nerves directly to skeletal muscle.
Knowing which level is damaged allows you to localize the lesion precisely.
Comparison Table
| Sign | UMN Lesion | LMN Lesion |
|---|---|---|
| Weakness | Yes | Yes |
| Muscle atrophy | None (mild disuse atrophy late) | Severe, early |
| Fasciculations | Absent | Common (spontaneous firing of denervated motor units) |
| Muscle tone | Increased - spastic | Decreased - flaccid |
| Deep tendon reflexes | Hyperactive | Hypoactive / absent |
| Babinski sign | Present (extensor plantar response) | Absent |
| Clonus | May be present | Absent |
| Distribution of weakness | Pyramidal / regional (e.g., arm flexors + leg extensors spared) | Distal or segmental (follows the nerve/root) |
(Source: Harrison's Principles of Internal Medicine 22E, Table 26-1; Neuroanatomy through Clinical Cases 3rd Ed, Table 6.4)
Key Features Explained
UMN Lesions
- Spasticity: Velocity-dependent increase in tone (catches when you move the limb quickly). Caused by damage to descending inhibitory pathways that travel alongside the corticospinal tract, leading to increased excitability of anterior horn motor neurons.
- Hyperreflexia: Exaggerated stretch reflexes (e.g., brisk knee jerk, ankle clonus).
- Babinski sign: Dorsiflexion of the great toe + fanning of other toes on stroking the lateral sole. Normal in infants (corticospinal tract not yet myelinated); pathological in adults.
- No significant atrophy: The LMN and muscle are intact; only mild disuse wasting develops over time.
- Pyramidal distribution: In the arm, flexors are relatively stronger (extensors weak); in the leg, extensors are relatively stronger (flexors weak) - producing the classic hemiplegic posture/gait.
- Important caveat: Acute UMN lesions (e.g., acute stroke, acute spinal cord injury / spinal shock) initially produce flaccid paralysis with decreased tone and decreased reflexes. Spasticity develops over hours to months as the cord adapts.
LMN Lesions
- Flaccid paralysis: Loss of all motor neuron influence on the muscle - no tone, no reflex arc.
- Atrophy: Early and severe; the muscle loses its trophic supply from the motor neuron.
- Fasciculations: Visible/palpable muscle twitches caused by spontaneous discharge of a denervated motor unit. A classic sign of LMN disease (e.g., ALS, radiculopathy).
- Areflexia: The reflex arc (LMN → muscle) is broken.
- Segmental/distal distribution: Weakness matches the territory of the affected nerve root, peripheral nerve, or anterior horn segment.
Pathophysiology Summary
Cerebral cortex
↓
[UMN] corticospinal tract
↓
Anterior horn (spinal cord) or cranial nerve nucleus
↓
[LMN] peripheral nerve
↓
Muscle
- Damage above the anterior horn cell = UMN signs
- Damage at or below the anterior horn cell = LMN signs
Special Situations
Facial Nerve (CN VII)
- UMN lesion (e.g., stroke): Contralateral lower face weakness only. The upper face is spared because it receives bilateral cortical input (both hemispheres contribute to forehead movement).
- LMN lesion (e.g., Bell's palsy): Entire ipsilateral face is weak - upper and lower.
Combined UMN + LMN (e.g., ALS)
- Amyotrophic Lateral Sclerosis destroys both anterior horn cells (LMN - atrophy, fasciculations) and corticospinal tracts (UMN - spasticity, hyperreflexia). This combination of fasciculations + brisk reflexes in a wasted limb is pathognomonic.
Spinal Cord Lesions
- At the level of injury: LMN signs (anterior horn cell damage)
- Below the level of injury: UMN signs (corticospinal tract interruption)
Quick Memory Aid
| UMN | LMN | |
|---|---|---|
| Tone | Up (spastic) | Down (flaccid) |
| Reflexes | Up | Down |
| Atrophy | No | Yes |
| Fasciculations | No | Yes |
| Babinski | Yes (+) | No (-) |
A simple mnemonic: UMN = UP (reflexes up, tone up) | LMN = DOWN + ATROPHY
Sources: Neuroanatomy through Clinical Cases, 3rd Ed (Blumenfeld), Table 6.4; Harrison's Principles of Internal Medicine 22E, Table 26-1; Ganong's Review of Medical Physiology, 26th Ed
Myocardial infarction
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive data from Robbins, Ganong, and Guyton. I have the morphology table content from line 585. Let me compile the full, well-structured answer now.
Myocardial Infarction (MI)
Definition
MI ("heart attack") is necrosis of cardiac muscle resulting from ischemia. The 2018 joint ESC/ACC/AHA definition: "presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- Myocyte dysfunction begins within 1-2 minutes of ischemia
- Irreversible cell death occurs after 20-40 minutes
- The infarct achieves its full extent in 3-6 hours - the critical therapeutic window
Pathogenesis
The typical sequence (in ~90% of cases):
- Atherosclerotic plaque disruption or erosion - exposes subendothelial collagen and necrotic plaque contents
- Platelet adhesion and aggregation - release of TXA₂, ADP, serotonin → further aggregation + vasospasm
- Coagulation cascade activation via exposed tissue factor → growing thrombus
- Complete coronary occlusion within minutes
In 10% of cases, transmural MI occurs without occlusive atherosclerosis - due to vasospasm, embolization from mural thrombus or valve vegetations, or small vessel disease (vasculitis, amyloid, sickle cell).
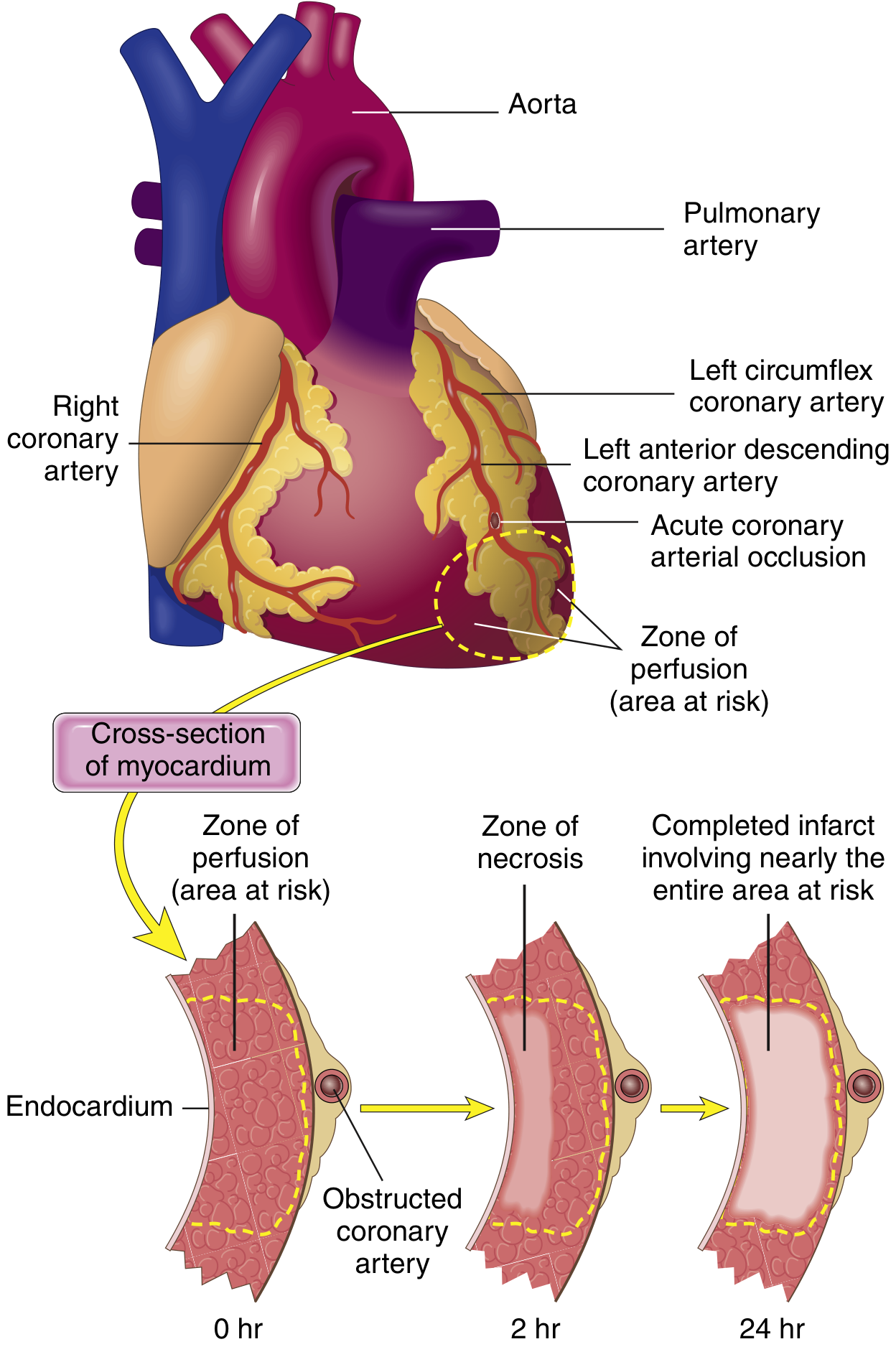
Patterns of Infarction
By depth
- Transmural - full thickness of ventricle wall; caused by epicardial vessel occlusion + thrombosis
- Subendocardial - inner one-third only; thrombus lysed before necrosis becomes transmural, or demand ischemia (tachycardia, hypotension, anemia) on a background of fixed stenosis
By vessel territory (Robbins)
| Artery occluded | % of MIs | Territory infarcted |
|---|---|---|
| LAD (proximal) | 40-50% | Anterior LV, anterior 2/3 of septum, apex |
| RCA (proximal) | 30-40% | RV, posterior/inferior LV |
| LCX | 15-20% | Lateral LV |
Subendocardial muscle is most vulnerable because: (1) it is the last area to receive blood from epicardial vessels, and (2) intramural pressure during systole compresses its vessels intensely.
Morphological Timeline (Robbins - Table 9.2)
| Time Frame | Gross Appearance | Microscopic Features |
|---|---|---|
| 0-12 hours | None visible (TTC stain shows pale area after 3 hrs) | None (earliest = wavy fibers at margins) |
| 12-24 hours | Red-blue discoloration (trapped blood) | Coagulative necrosis; pyknotic nuclei; "wavy fibers" |
| 1-3 days | Pale, yellow-tan | Loss of nuclei + striations; neutrophilic infiltrate |
| 3-7 days | Pale/yellow, soft center | Abundant neutrophils; macrophage infiltration begins |
| 5-10 days | Yellow-tan, maximally soft (risk of rupture) | Macrophages and granulation tissue at margins |
| 1-2 weeks | Hyperemic (red) rim of granulation tissue | Granulation tissue with new vessels + collagen |
| 2-8 weeks | Gray-white scar progressively forming | Fibrosis advancing inward |
| >2 months | White, firm fibrous scar | Dense collagen scar - cannot date the infarct |
Healing progresses from border to center. Large infarcts heal more slowly. Old scars (8 weeks or 10 years old) look identical.
ECG Changes
Three electrical events in infarcted cells produce ECG changes:
| Defect in Infarcted Cells | Current Flow | ECG Change (leads over infarct) |
|---|---|---|
| Rapid repolarization (K⁺ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential | Into infarct | TQ depression (recorded as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
Serial ECG changes in anterior MI:
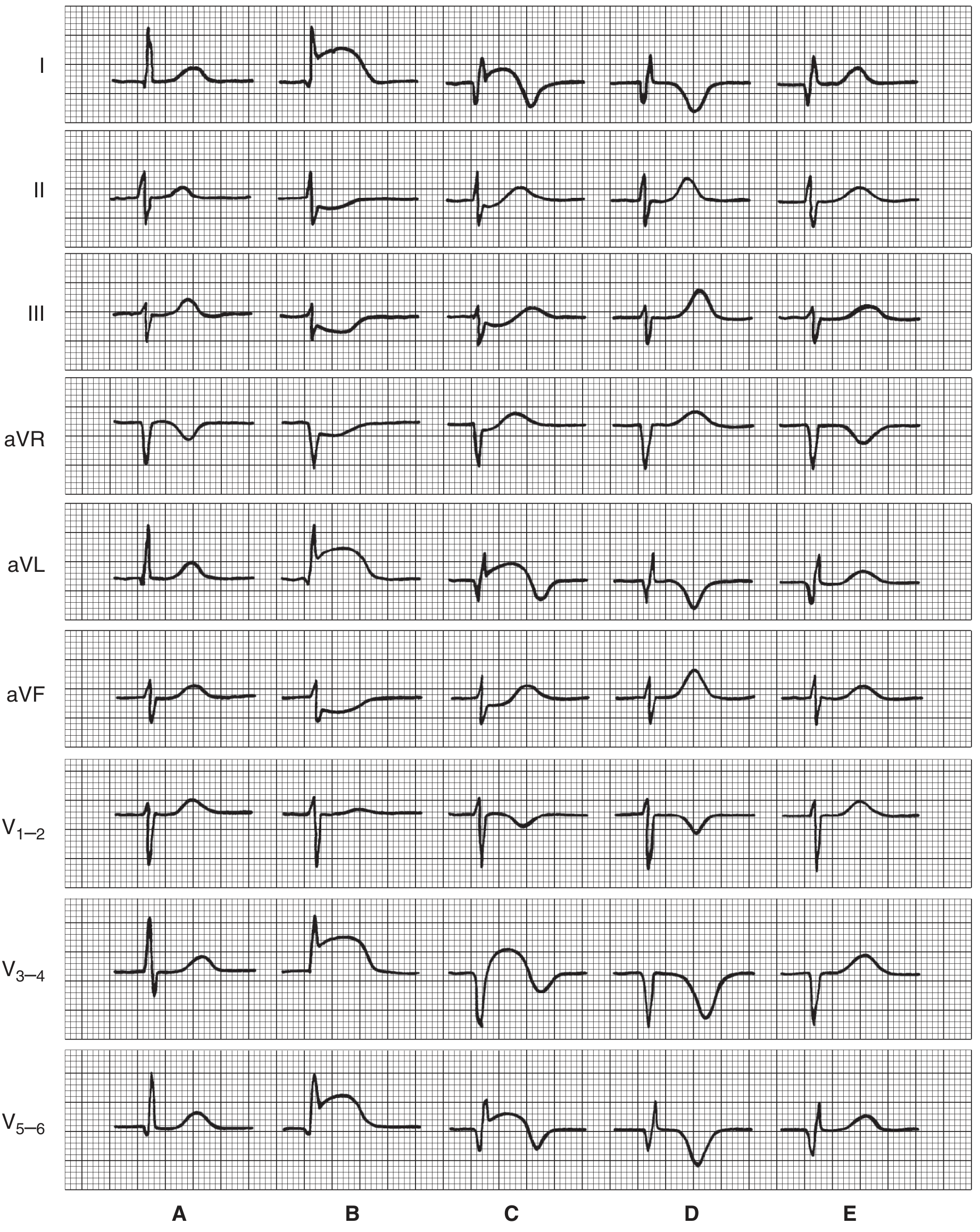
- (A) Normal
- (B) Very early (hours): ST elevation in I, aVL, V3-5; reciprocal ST depression in II, III, aVF
- (C) Hours to days: Q waves appear; ST changes persist; T-wave inversion begins
- (D) Days to weeks: Deep Q/QS; ST near-baseline; deep T inversion
- (E) Weeks to months: Q waves remain (permanent); T waves normalize ("chronic" pattern)
Reciprocal changes: Leads on the opposite side of the heart show ST depression.
Non-Q-wave infarcts: Tend to be less transmural but carry a high risk of subsequent reinfarction.
Biomarkers
- Troponin I / Troponin T - gold standard; most sensitive and specific; rise within 3-4 hours, peak at ~24 hrs (cTnI) or ~48 hrs (cTnT), remain elevated 7-10+ days
- CK-MB - rises within 4-6 hours, peaks at ~24 hours, normalizes by 48-72 hours; useful for detecting reinfarction
- Myoglobin - earliest to rise (~1-2 hours) but not cardiac-specific
- Myocyte membrane disruption is the earliest detectable event, allowing intracellular macromolecules to leak into the interstitium and vasculature
Complications
(Robbins & Guyton)
| Complication | Details |
|---|---|
| Arrhythmias | Most common cause of death (80-90% of ischemic cardiac deaths = VF); reentry arrhythmias in first 30 min, automaticity-based arrhythmias after 12 hours |
| Cardiogenic shock | Decreased cardiac output; worsened by systolic stretch - infarcted wall bulges outward during systole, wasting pumping force |
| Pulmonary edema | Failure of LV → backing up into pulmonary circulation |
| Ventricular free wall rupture | Peak risk day 5-10 (maximal softening); causes tamponade; ~1-5% of MIs |
| Ventricular septal defect | Rupture of infarcted septum |
| Papillary muscle rupture | Acute mitral regurgitation; severe hemodynamic compromise |
| Left ventricular aneurysm | Fibrotic bulge; persistent ST elevation on ECG; risk of mural thrombus |
| Mural thrombus | On infarcted endocardium; risk of systemic embolization |
| Pericarditis | Dressler syndrome (autoimmune) 2-10 weeks post-MI; or early infarct pericarditis |
| Ischemic cardiomyopathy | Progressive HF from accumulated ischemic injury |
Reperfusion and Reperfusion Injury
The therapeutic goal is "time is myocardium" - restore perfusion via thrombolysis (tPA), PCI (angioplasty/stenting), or CABG. However, reperfusion causes its own injury:
- Mitochondrial dysfunction - ischemia alters membrane permeability → swelling → rupture → apoptosis
- Calcium overload - increased intracellular Ca²⁺ → myofibril hypercontracture → cell death
- Free radical burst - O₂⁻, H₂O₂, hydroxyl radicals damage membrane proteins and lipids within minutes of reperfusion
- No-reflow phenomenon - leukocyte aggregation occludes microvasculature, preventing perfusion even after vessel opening
- Stunned myocardium - viable but non-contractile myocytes post-reperfusion; may persist for days
Key Management Principles
(Ganong / Robbins)
- Aspirin + P2Y12 inhibitor (dual antiplatelet) - inhibit further platelet aggregation
- Primary PCI - reperfusion of choice within 90 minutes of first medical contact
- Thrombolytics - if PCI unavailable within 120 min
- Beta-blockers - reduce myocardial oxygen demand, limit infarct size, anti-arrhythmic
- ACE inhibitors - reduce adverse remodeling and mortality
- Statins - plaque stabilization and LDL reduction
- Anticoagulation - heparin during PCI; prevents mural thrombus
- Riluzole - in ALS-associated motor neuron disease involving glutamate excitotoxicity (separate indication)
- Spasticity from ischemic injury can be treated with baclofen or tizanidine
Sources: Robbins & Kumar Basic Pathology (Robbins Pathology), Ch. 9; Guyton and Hall Textbook of Medical Physiology, Ch. 21; Ganong's Review of Medical Physiology 26th Ed, Ch. 29
Abnormal uterine bleeding
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive data from Robbins, Sabiston, Rosen's, and Berek & Novak. Let me compile the full structured answer.
Abnormal Uterine Bleeding (AUB)
Definition and Normal Parameters
AUB is any bleeding from the uterus that differs in frequency, duration, regularity, or volume from a normal menstrual cycle.
Normal menstrual parameters:
- Cycle length: 24-38 days
- Duration of flow: 4-8 days (>7 days = abnormal)
- Average blood loss: 35 mL per cycle; >80 mL = abnormal (heavy menstrual bleeding)
The term "dysfunctional uterine bleeding (DUB)" is no longer recommended. The current standard term is AUB with PALM-COEIN classification (FIGO 2011, endorsed by ACOG).
PALM-COEIN Classification (FIGO 2011)
The PALM-COEIN system divides AUB causes into structural and non-structural:
PALM - Structural Causes
(diagnosed by imaging or histology)
| Letter | Cause | Key Features |
|---|---|---|
| P | Polyp | Endometrial gland/stroma overgrowth; sessile or pedunculated; feeder vessel on Doppler; mostly benign but ~5% malignant transformation; associated with tamoxifen use, obesity, PCOS |
| A | Adenomyosis | Endometrial glands/stroma within the myometrium; causes heavy, painful periods; diagnosed on MRI or ultrasound |
| L | Leiomyoma (fibroid) | Most common benign gynecologic tumor; affects up to 70% of women by age 50; more prevalent and severe in Black women; submucosal fibroids (Types 0-2) are most likely to cause AUB - increased endometrial surface area prevents adequate vessel compression |
| M | Malignancy & Hyperplasia | Must always be excluded, especially postmenopausal; hyperplasia with atypia (EIN) = PTEN mutations, precancerous |
COEIN - Non-structural Causes
(medical/functional diagnoses)
| Letter | Cause | Key Features |
|---|---|---|
| C | Coagulopathy | Von Willebrand disease most common; screen if: heavy bleeding since menarche, family history, multiple system bleeding, medication-related |
| O | Ovulatory dysfunction | ~50% of cases of excess menstruation; includes anovulation (no corpus luteum → no progesterone → unopposed estrogen → unstable endometrium); PCOS is the leading cause in reproductive years |
| E | Endometrial | Primary endometrial disorder of hemostasis (e.g., reduced prostaglandin synthesis, endometritis) |
| I | Iatrogenic | Medications: anticoagulants, SSRIs, antipsychotics, hormonal therapy, copper IUD, tamoxifen |
| N | Not otherwise classified | Rare/poorly defined entities (e.g., arteriovenous malformations) |
Causes by Age Group (Robbins)
| Age Group | Common Causes |
|---|---|
| Prepuberty | Precocious puberty (hypothalamic/pituitary/ovarian origin) |
| Adolescence | Anovulatory cycles, coagulopathy (especially Von Willebrand disease) |
| Reproductive age | Pregnancy complications (ectopic, abortion, trophoblastic disease), anatomic lesions (fibroids, polyps, adenomyosis, hyperplasia), anovulatory DUB |
| Perimenopause | Anovulatory cycles, inadequate luteal phase, anatomic lesions |
| Postmenopause | Endometrial atrophy (most common), carcinoma, hyperplasia, polyps - any postmenopausal bleeding must be considered abnormal and investigated |
Pathophysiology of Key Causes
Anovulatory Bleeding (most common, ~50% of cases)
- No ovulation → no corpus luteum → no progesterone produced
- Unopposed estrogen → endometrium proliferates excessively → becomes unstable → breaks down irregularly and unpredictably
- Most common at: menarche and perimenopause (HPO axis fluctuation), and in PCOS
Fibroids (Leiomyomata)
- Submucosal fibroids are most problematic: increased endometrial surface area + disruption of uterine contraction mechanism → vessel compression fails → heavy bleeding
Endometrial Hyperplasia
- Estrogen excess (obesity, PCOS, granulosa cell tumors, exogenous estrogens) → endometrial gland proliferation
- Without atypia: 1-3% risk of progression to carcinoma
- With atypia (EIN): associated with PTEN mutations; considered a carcinoma precursor; requires hysterectomy (or high-dose progestins if fertility preservation needed)
Workup / Investigation
All patients:
- Detailed menstrual history and pelvic exam
- Pregnancy test (urine/serum β-hCG) - exclude first
- CBC (anemia assessment)
- Thyroid function tests (hypothyroidism causes menorrhagia)
- Prolactin level (hyperprolactinemia disrupts GnRH → LH/FSH reduction → anovulation)
- Cervical cancer screening (if not current)
- STI screening
- Pelvic ultrasound - first-line imaging
Coagulation screen if:
- Heavy bleeding since menarche
- Family history of coagulopathy
- Medication-associated bleeding
- Signs of systemic bleeding (epistaxis, easy bruising)
Endometrial biopsy (EMB) - indicated in:
- Age ≥45 years with AUB (including intermenstrual bleeding)
- Age <45 with risk factors: obesity, unopposed estrogen, ovulatory dysfunction, persistent/refractory AUB, elevated familial cancer risk (Lynch syndrome, Cowden syndrome)
Hysteroscopy: Gold standard for evaluating intracavitary lesions (polyps, submucosal fibroids); allows direct visualization and biopsy.
Management
Medical (First-line for non-structural causes)
| Drug | Mechanism / Use | Dosing |
|---|---|---|
| Combined OCP | Regulate cycle, reduce flow | Monophasic pill TID × 7 days or BID × 5 days then QD |
| Progestin-only (MPA) | Stabilize endometrium; used if estrogen contraindicated | 20 mg TID × 7 days |
| IV Conjugated Estrogen | Acute hemostasis in severe bleeding | 25 mg IV q4-6h up to 24 hours |
| NSAIDs (ibuprofen, mefenamic acid, naproxen) | Inhibit prostaglandins → reduce flow by 20-50%; also analgesia | Ibuprofen 200-400 mg TID/QID × 5 days |
| Tranexamic acid | Antifibrinolytic; reduces heavy menstrual bleeding | 1.3 g PO q6-8h × up to 5 days |
| Levonorgestrel IUD (Mirena) | Local progesterone → endometrial atrophy; reduces flow by ~90%; also treats dysmenorrhea | Long-term; first-line for ovulatory AUB |
| GnRH agonists | Suppress HPO axis → temporary menopause; used pre-surgery to shrink fibroids | Short-term only (bone loss) |
Surgical
| Procedure | Indication |
|---|---|
| Endometrial ablation | Destroys endometrial lining; good for women who have completed childbearing; ~29% require hysterectomy by 5 years |
| Hysteroscopic polypectomy | Uterine polyps |
| Myomectomy | Fertility-preserving fibroid removal (open/laparoscopic/hysteroscopic depending on type) |
| Uterine artery embolization (UAE) | Fibroids; less invasive than hysterectomy; ~31% eventually need hysterectomy |
| Hysterectomy | Definitive; reserved for failure/intolerance of medical therapy, malignancy, or adenomyosis; endometrial sampling must be done first |
Special Scenarios
Postmenopausal Bleeding
- Always abnormal - requires thorough evaluation
- Most common cause: endometrial atrophy (not carcinoma)
- Most feared cause: endometrial carcinoma (endometrioid type in 80% - estrogen-related; serous type in 15% - atrophy-related, more aggressive)
- Workup: pelvic ultrasound (endometrial thickness), EMB
Acute Heavy Bleeding (Emergency Department)
- IV conjugated estrogen for rapid hemostasis
- High-dose OCP regimens
- Tranexamic acid
- If hemodynamically unstable: resuscitation, urgent gynaecology consult, possible surgical intervention
Sources: Robbins & Kumar Basic Pathology, Table 17.2; Sabiston Textbook of Surgery 11th Ed, Ch. 120; Rosen's Emergency Medicine 9th Ed, Table 30.1; Berek & Novak's Gynecology; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.