Fats and lipids metabolism in biochemistry
Reading File
Reading File
Finding Sources
Running Command
python3 -c " from pypdf import PdfReader reader = PdfReader('/home/daytona/works…
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
I now have all the content I need. Let me compile the comprehensive response based on the student's PDF notes and the textbook sources.
Fats & Lipid Metabolism - Complete Biochemistry Notes
Based on your attached notes + Guyton & Hall Medical Physiology + Harper's Illustrated Biochemistry
1. Classification of Lipids
Lipids are heterogeneous molecules insoluble in water. The main classes are:
| Class | Examples |
|---|---|
| Simple lipids (triesters) | Triglycerides (TAG), Sterols |
| Complex/Conjugated lipids | Phospholipids, Glycolipids, Lipoproteins, Sulfolipids |
| Derived lipids | Fatty acids, Eicosanoids, Steroids |
2. Fatty Acids (FA)
Classification by Chain Length
| Type | Carbon atoms |
|---|---|
| SCFA (Short chain) | C2 - C6 |
| MCFA (Medium chain) | C8 - C12 |
| LCFA (Long chain) | C14 - C18 (e.g., Palmitate C16:0, Stearate C18:0) |
| VLCFA (Very long chain) | C20+ |
Essential vs. Non-essential Fatty Acids
- Essential FA (EFA) - must come from diet: Linoleic acid (18:2, ω-6) and α-Linolenic acid (18:3, ω-3)
- Semi-essential: Arachidonic acid (20:4, ω-6), DHA (22:6, ω-3), EPA (20:5, ω-3)
- Deficiency of EFA → Phrynoderma (toad skin)
Saturated vs. Unsaturated
- Saturated (no double bonds): Palmitic acid (16:0), Stearic acid (18:0)
- Monounsaturated: Oleic acid (18:1, ω-9)
- Polyunsaturated (PUFA): Linoleic, Linolenic, ARA, DHA, EPA
Omega (ω) Numbering System
The omega carbon is the farthest carbon from the carboxyl group. Double bond position counted from that end.
- Example: 2,4-dienoic hexanoic acid → ω-3 series
Trans Fatty Acids (TFA)
- Elaidic acid (trans form of oleic) found in vanaspati (partially hydrogenated vegetable fat)
- TFA ↑ LDL cholesterol, ↑ CVD risk
ω-3 vs. ω-6 Health Effects
| ω-3 (DHA, EPA, α-Linolenic) | ω-6 (Arachidonic, Linoleic) |
|---|---|
| Cardioprotective | Arachidonic acid → Prostaglandins (pro-inflammatory) |
| Rich in fish oil, flaxseed, evening primrose | Sunflower oil, corn oil |
| DHA essential for brain development (mother's milk) | ↑ risk of CVD at excess |
3. Phospholipids
Phospholipids are conjugated lipids (lipid + ionized part).
Glycerophospholipids (alcohol = glycerol)
| Name | Also called | Key role |
|---|---|---|
| Phosphatidylcholine | Lecithin | Lung surfactant (as DPPC - Dipalmitoyl Phosphatidylcholine) |
| Phosphatidylethanolamine | Cephalin | Platelet aggregation |
| Phosphatidylserine | - | Identifies apoptotic cells (flips to outer leaflet) |
| Phosphatidylinositol | - | Cell signalling (PIP2 → IP3 + DAG) |
| Cardiolipin | Diphosphatidylglycerol | Inner mitochondrial membrane; deficiency → Barth's syndrome |
DPPC (Lecithin) is the main lung surfactant. Deficiency → Respiratory Distress Syndrome (RDS) in neonates. The Lecithin:Sphingomyelin (L:S) ratio tests fetal lung maturity; L:S ≥ 2 indicates lung maturity.
Sphingophospholipids (alcohol = sphingosine)
- Sphingomyelin - lines the myelin sheath of neurons
4. Sphingolipidoses
These are lysosomal storage disorders caused by deficiency of specific hydrolase enzymes, leading to accumulation of the respective sphingolipid. All are Autosomal Recessive (AR) except Fabry's disease (X-linked).
| Disease | Enzyme deficient | Accumulated substrate | Key features |
|---|---|---|---|
| Niemann-Pick | Sphingomyelinase | Sphingomyelin | Zebra body (membrane-bound) inclusions |
| Gaucher's | β-Glucocerebrosidase | Glucocerebroside | Erlenmeyer flask femur deformity, "crumpled tissue paper" Gaucher cells, no cherry-red spot |
| Tay-Sachs | Hexosaminidase A | GM2 ganglioside | Cherry-red spot on macula, hyperacusis |
| Krabbe's | Galactocerebrosidase | Galactocerebroside | Neuropathy |
| Metachromatic Leukodystrophy | Arylsulfatase A | Sulfatide | Mimics rheumatoid arthritis |
| Fabry's (X-linked) | α-Galactosidase A | Globotriaosylceramide | Angiokeratoma, febrile episodes |
5. Lipoproteins
Lipoproteins transport lipids through the bloodstream. They consist of a hydrophobic lipid core surrounded by amphipathic phospholipids, cholesterol, and apolipoproteins.
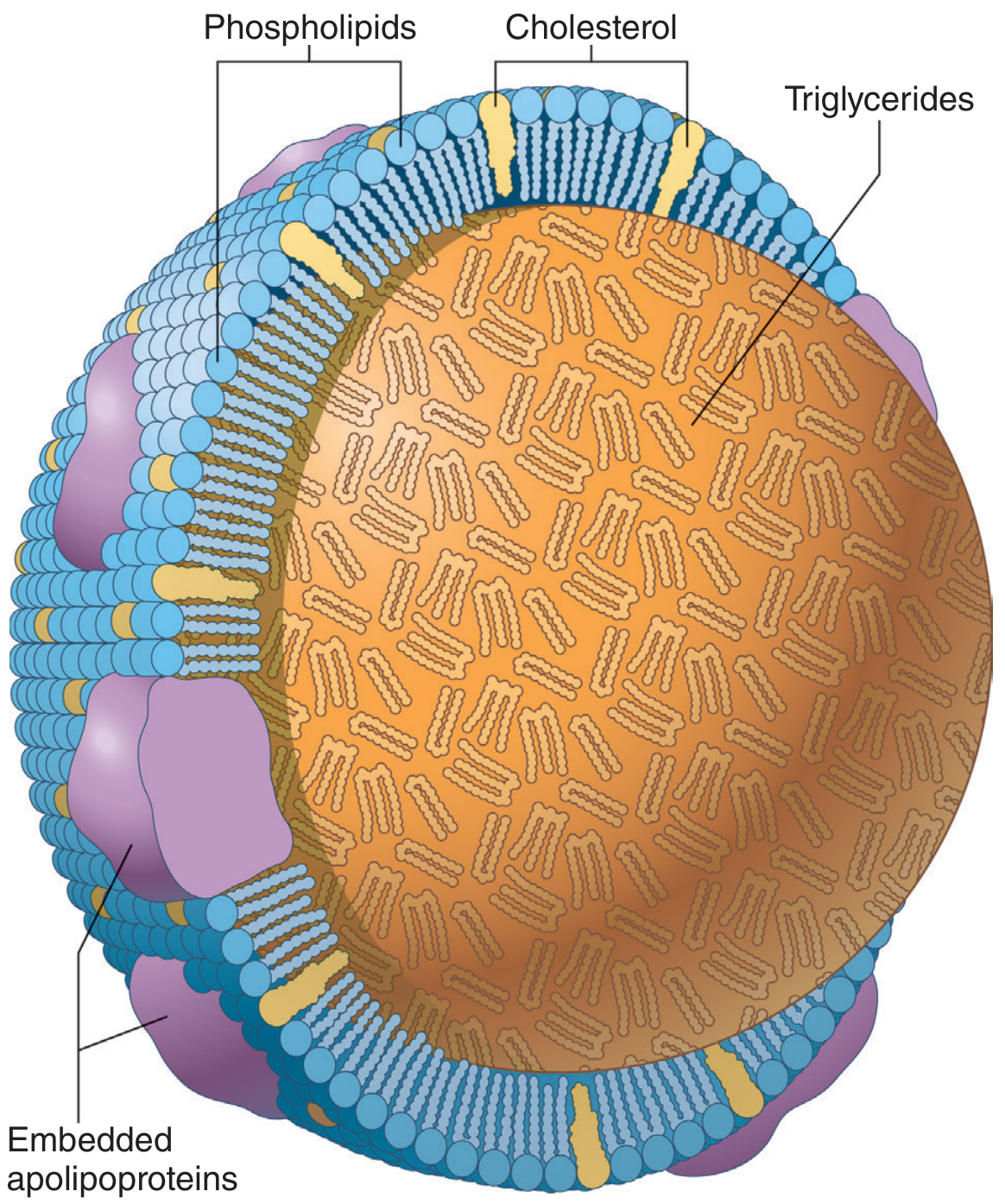
Types (by density, ultracentrifugation)
| Lipoprotein | Main Lipid | Apolipoprotein | Function |
|---|---|---|---|
| Chylomicrons | Max TG (dietary) | ApoB-48, ApoC-II, ApoE | Transport dietary (exogenous) TG from gut → tissues |
| VLDL | High TG (endogenous) | ApoB-100, ApoC-II, ApoE | Transport liver-made TG → adipose/muscle |
| IDL | TG + Cholesterol | ApoB-100, ApoE | Intermediate - taken up by liver or → LDL |
| LDL | Max Cholesterol | ApoB-100 | "Bad cholesterol" - delivers cholesterol to tissues |
| HDL | Max Protein | ApoA-I | "Good cholesterol" - reverse cholesterol transport |
Electrophoresis order (anode to origin): HDL (α) → LDL (β) → IDL (pre-β) → VLDL → Chylomicrons (origin)
Key Apolipoproteins
- ApoB-48 - chylomicrons only (made in intestine via RNA editing of ApoB-100 mRNA)
- ApoB-100 - VLDL, IDL, LDL (made in liver)
- ApoC-II - activates Lipoprotein Lipase (LPL)
- ApoE - receptor-mediated endocytosis (hepatic uptake)
- ApoA-I - activates LCAT (Lecithin Cholesterol Acyltransferase)
Enzyme: LCAT (Lecithin Cholesterol Acyltransferase)
- Esterifies free cholesterol → cholesteryl ester (in HDL)
- Deficiency → Norum disease (fish-eye disease)
Enzyme: Acid Lipase
- Deficiency → Wolman's disease (watery green diarrhea, vomiting, adrenal calcification)
6. Hyperlipoproteinemias (Fredrickson Classification)
| Type | Elevated lipoprotein | Defect |
|---|---|---|
| I | Chylomicrons | LPL deficiency |
| IIa | LDL | LDL receptor defect (Familial Hypercholesterolemia, AD) |
| III | IDL | ApoE defect |
| IV | VLDL | VLDL overproduction |
| V | Chylomicrons + VLDL | Mixed |
Hypolipoproteinemias:
- Abetalipoproteinemia - no ApoB-containing particles (can't absorb fat)
- Tangier's disease - HDL deficiency (ApoA-I deficiency)
7. Cholesterol Metabolism
Cholesterol is amphipathic (hydrophobic ring + hydrophilic OH). Sources: diet, bile acids, de novo synthesis, vitamin D, steroid hormones.
De Novo Cholesterol Synthesis (in Cytosol)
Precursor: Acetyl-CoA
Acetyl-CoA → Acetoacetyl-CoA → HMG-CoA → Mevalonate → ... → Cholesterol
- Rate-Limiting Enzyme (RLE): HMG-CoA Reductase
- Inhibited by Statins (competitive, suicide inhibition)
- Active in dephosphorylated form
- Activated by Insulin and Thyroxine
RLE of bile acid synthesis: 7α-Hydroxylase
Eicosanoids (Derived from Arachidonic Acid)
- COX pathway → Prostaglandins, Prostacyclins, Thromboxanes
- Inhibited by Aspirin (irreversible/suicide inhibition)
- Inhibited by NSAIDs (reversible)
- LOX pathway → Leukotrienes
8. Lipogenesis (De Novo Fatty Acid Synthesis)
- Process: Anabolic, occurs in cytoplasm (cytosol)
- State: Well-fed (high insulin)
- Precursor: Acetyl-CoA
- Product: Palmitic acid (16:0) after 7 cycles
Steps
- Citrate shuttle moves Acetyl-CoA from mitochondria to cytosol (via citrate → OAA + Acetyl-CoA by ATP-citrate lyase)
- Rate-Limiting Step: Acetyl-CoA + CO₂ → Malonyl-CoA by Acetyl-CoA Carboxylase (ACC)
- Requires Biotin as cofactor
- Activated by insulin (dephosphorylated, active form)
- Inhibited by glucagon/epinephrine (phosphorylated, inactive form)
Fatty Acid Synthase (FAS) Complex
FAS has two key -SH groups:
- Cys-SH - accepts Acetyl-CoA (condensing unit)
- Pan-SH (Phosphopantetheine) - accepts Malonyl-CoA (extending unit)
4 reactions per cycle (add 2 carbons each cycle):
- Condensation (Ketoacyl synthase)
- Reduction (Ketoacyl reductase) - uses NADPH
- Dehydration (Dehydratase)
- Reduction (Enoyl reductase) - uses NADPH
After 7 cycles → Palmitate (16:0)
Regulation: Lipogenesis vs. Lipolysis
| Lipogenesis | Lipolysis | |
|---|---|---|
| Process | Anabolic | Catabolic |
| Location | Cytoplasm | Mitochondria |
| RLE | Acetyl-CoA Carboxylase | Hormone-sensitive lipase |
| Active state | Fed (insulin↑) | Fasted/stress (glucagon/adrenaline↑) |
| Key molecule | Malonyl-CoA (inhibits CPT-I → prevents β-oxidation) | Fatty acids → Acyl-CoA |
Malonyl-CoA acts as the key switch: when lipogenesis is ON, Malonyl-CoA inhibits CPT-I and blocks β-oxidation, preventing futile cycling.
9. β-Oxidation (Lipolysis / Catabolism of Fatty Acids)
- Process: Catabolic, occurs in mitochondria
- State: Fasting, starvation, stress
Steps
Step 1 - Activation: FA + CoA → Acyl-CoA (costs 2 ATP = 1 ATP→ AMP + PPi, equivalently 2 ATP equivalents)
Step 2 - Transport (Carnitine Shuttle):
- Long-chain FA (LCFA) cannot cross inner mitochondrial membrane - require carnitine
- CPT-I (outer membrane) - transfers acyl group to carnitine
- CPT-II (inner membrane) - regenerates Acyl-CoA inside matrix
- SCFA and MCFA do NOT require carnitine (cross freely)
- VLCFA are first oxidized in peroxisomes (α-oxidation, require PTS - Peroxisomal Targeting Sequence)
Step 3 - β-Oxidation (4 repeating steps):
- Oxidation (FAD → FADH₂) - Acyl-CoA Dehydrogenase
- Hydration (water added)
- Oxidation (NAD⁺ → NADH)
- Thiolytic cleavage (releases Acetyl-CoA, shortens chain by 2C)
Energetics of Palmitate (C16:0)
- Cycles needed: 7 cycles (n/2 - 1 = 8-1 = 7)
- Products: 8 Acetyl-CoA + 7 NADH + 7 FADH₂
- Net ATP = ~106 ATP (subtract 2 ATP for activation)
Odd-Chain FA
- Produces Acetyl-CoA + Propionyl-CoA (3-carbon)
- Propionyl-CoA → Methylmalonyl-CoA → Succinyl-CoA (enters TCA) - requires Vitamin B12
α-Oxidation (Peroxisomal)
- Oxidizes Phytanic acid (from green leafy vegetables, milk)
- Enzyme: α-Phytanoyl-CoA hydroxylase
- Deficiency → Refsum's disease (retinitis pigmentosa, neuropathy, loss of hearing)
ω-Oxidation (ER microsomes)
- Minor pathway for medium-chain FA
Peroxisomal Disorders
- Mutation in PTS → VLCFA accumulate → Zellweger syndrome (cerebrohepatorenal syndrome) - mongoloid faces, epicanthal folds, "ghosts of peroxisomes"
- MCAD deficiency (Medium Chain Acyl-CoA Dehydrogenase deficiency) → SIDS (Sudden Infant Death Syndrome), ketotic hypoglycemia
10. Ketogenesis
Ketone bodies are produced in the liver from Acetyl-CoA, especially during fasting/starvation when oxaloacetate is diverted to gluconeogenesis.
Three Ketone Bodies
| Ketone body | Normal blood conc. | Nitroprusside test | β-OHB test |
|---|---|---|---|
| Acetoacetate | 1 mg/mL | + (purple ring) | Negative |
| β-Hydroxybutyrate (β-OHB) | 2 mg/mL | Negative | + |
| Acetone | Trace | + (volatile, breath odor) | Negative |
Normal total: ~1 mg/mL. In DM/starvation: 6:1 ratio of β-OHB:Acetoacetate.
Pathway of Ketogenesis (in liver mitochondria)
2 Acetyl-CoA → Acetoacetyl-CoA → HMG-CoA (via HMG-CoA Synthase - mitochondrial isoform) → Acetoacetate → β-OHB / Acetone
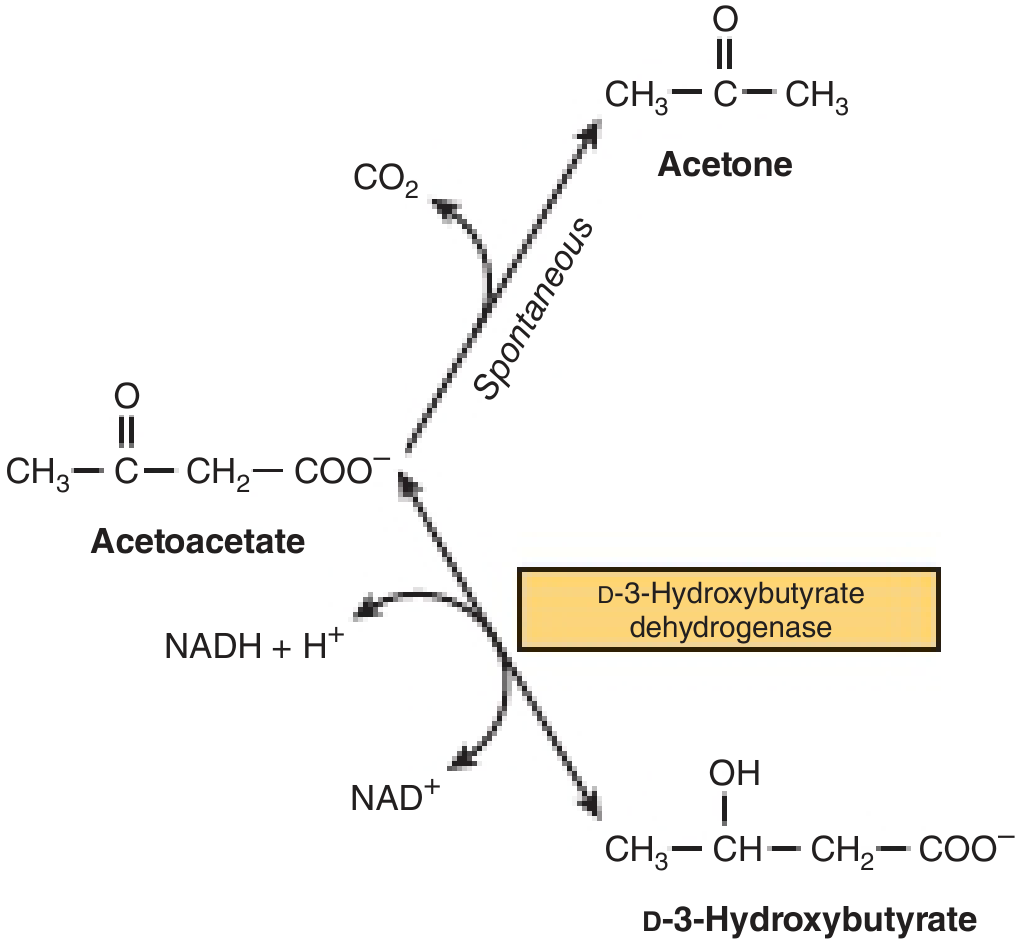
- RLE of Ketogenesis: HMG-CoA Synthase (mitochondrial)
- RLE of Cholesterol synthesis: HMG-CoA Reductase (cytosolic)
Organs That CANNOT Use Ketone Bodies
- RBCs - lack mitochondria (no Succinyl-CoA thiophorase/oxoacid CoA transferase)
- Liver - lacks the enzyme needed to utilize them (succinyl-CoA:3-ketoacid CoA transferase)
Brain uses ketone bodies preferentially during prolonged starvation
Three Key Regulation Points of Ketogenesis (Harper's)
- Lipolysis in adipose - FFA release (regulated by HSL)
- CPT-I - entry of FA into mitochondria (inhibited by Malonyl-CoA)
- Partition between esterification vs. oxidation in liver - determined by substrate availability
11. Comparison: β-Oxidation vs. Fatty Acid Synthesis
| Feature | β-Oxidation | Fatty Acid Synthesis |
|---|---|---|
| Location | Mitochondria | Cytoplasm |
| Process | Catabolic | Anabolic |
| Acyl carrier | CoA | ACP (part of FAS) |
| 2C unit | Released as Acetyl-CoA | Added as Malonyl-CoA (3C → 2C) |
| Coenzymes | FAD, NAD⁺ | NADPH (×2 per cycle) |
| Transport | Carnitine shuttle | Citrate shuttle |
| RLE | CPT-I (regulation) | Acetyl-CoA Carboxylase |
| Biotin | Not used | Required (for ACC) |
12. Key Enzymes and Their Clinical Significance
| Enzyme | Reaction | Clinical |
|---|---|---|
| Lipoprotein lipase (LPL) | Hydrolyzes TG in chylomicrons/VLDL | Deficiency → Type I hyperlipoproteinemia |
| HMG-CoA Reductase | Mevalonate synthesis (RLE cholesterol) | Target of statins |
| HMG-CoA Synthase (mito) | HMG-CoA in ketogenesis (RLE) | - |
| Acetyl-CoA Carboxylase | Malonyl-CoA synthesis (RLE lipogenesis) | Regulated by insulin/glucagon |
| CPT-I | FA entry into mitochondria | Inhibited by Malonyl-CoA |
| 7α-Hydroxylase | RLE of bile acid synthesis | - |
| LCAT | Cholesterol esterification in HDL | Deficiency → Norum disease |
| Acid lipase | Lysosomal cholesterol ester hydrolysis | Deficiency → Wolman's disease |
| Sphingomyelinase | Sphingomyelin hydrolysis | Deficiency → Niemann-Pick |
| β-Glucocerebrosidase | Glucocerebroside hydrolysis | Deficiency → Gaucher's |
| Hexosaminidase A | GM2 ganglioside hydrolysis | Deficiency → Tay-Sachs |
13. Clinical Tests for Ketone Bodies
- Rothera's test - detects acetoacetate and acetone → purple ring at junction
- Nitroprusside test - same principle
- β-OHB specific test - enzymatic (Acetest strips negative for β-OHB)
- Acetone is volatile → exhaled as fruity breath
Sources: Guyton and Hall Textbook of Medical Physiology, p. 842-844 | Harper's Illustrated Biochemistry 32nd Ed., pp. 232-235 | Your attached handwritten notes (28 pages)
This is a shared conversation. Sign in to Orris to start your own chat.