Explain apoptosis, it's mechanism
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
I now have comprehensive content from Robbins Cotran Pathologic Basis of Disease and other sources. Let me compile a thorough, well-organized answer.
Apoptosis — Definition and Mechanism
Apoptosis (Greek: "falling off," as leaves from a tree) is a form of programmed cell death in which cells activate intrinsic enzymes that systematically degrade their own nuclear DNA and cytoplasmic proteins. Unlike necrosis, it is an energy-dependent, organized process that clears unwanted cells without triggering inflammation.
Key Morphologic Features
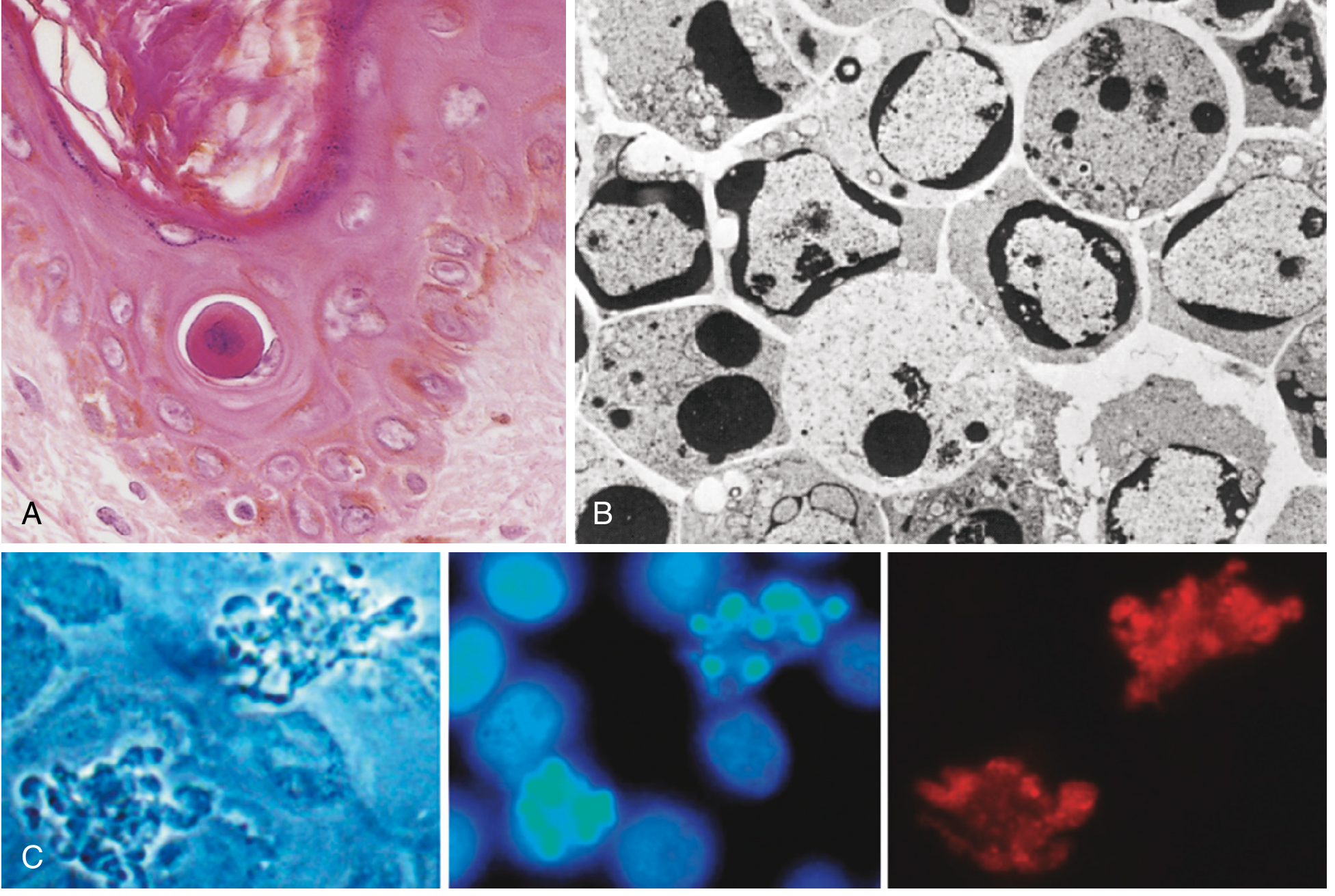
| Feature | Description |
|---|---|
| Cell shrinkage | Cell volume decreases; cytoplasm becomes dense and eosinophilic (opposite of necrosis) |
| Chromatin condensation | Chromatin aggregates peripherally under the nuclear membrane — the hallmark of apoptosis |
| Nuclear fragmentation | Nucleus breaks into 2 or more fragments |
| Membrane blebbing | Surface blebbing followed by fragmentation into apoptotic bodies (membrane-bound, contain organelles ± nuclear fragments) |
| Phagocytosis | Apoptotic bodies expose "eat-me" signals (e.g., phosphatidylserine) → rapidly engulfed by macrophages |
| No inflammation | Cell contents never leak into surrounding tissue, so no inflammatory response is triggered |
Central Executioners — Caspases
Apoptosis is mediated by caspases — cysteine proteases that cleave substrates after aspartic acid residues. They exist as inactive procaspases and are activated by:
- Enzymatic cleavage by another caspase
- Focal autocatalytic accumulation of inactive forms
The process has two phases:
- Initiation phase — specific initiator caspases (caspase-8 or -9) become active
- Execution phase — executioner caspases (caspase-3, -6, -7) mediate cellular fragmentation
Two Main Pathways
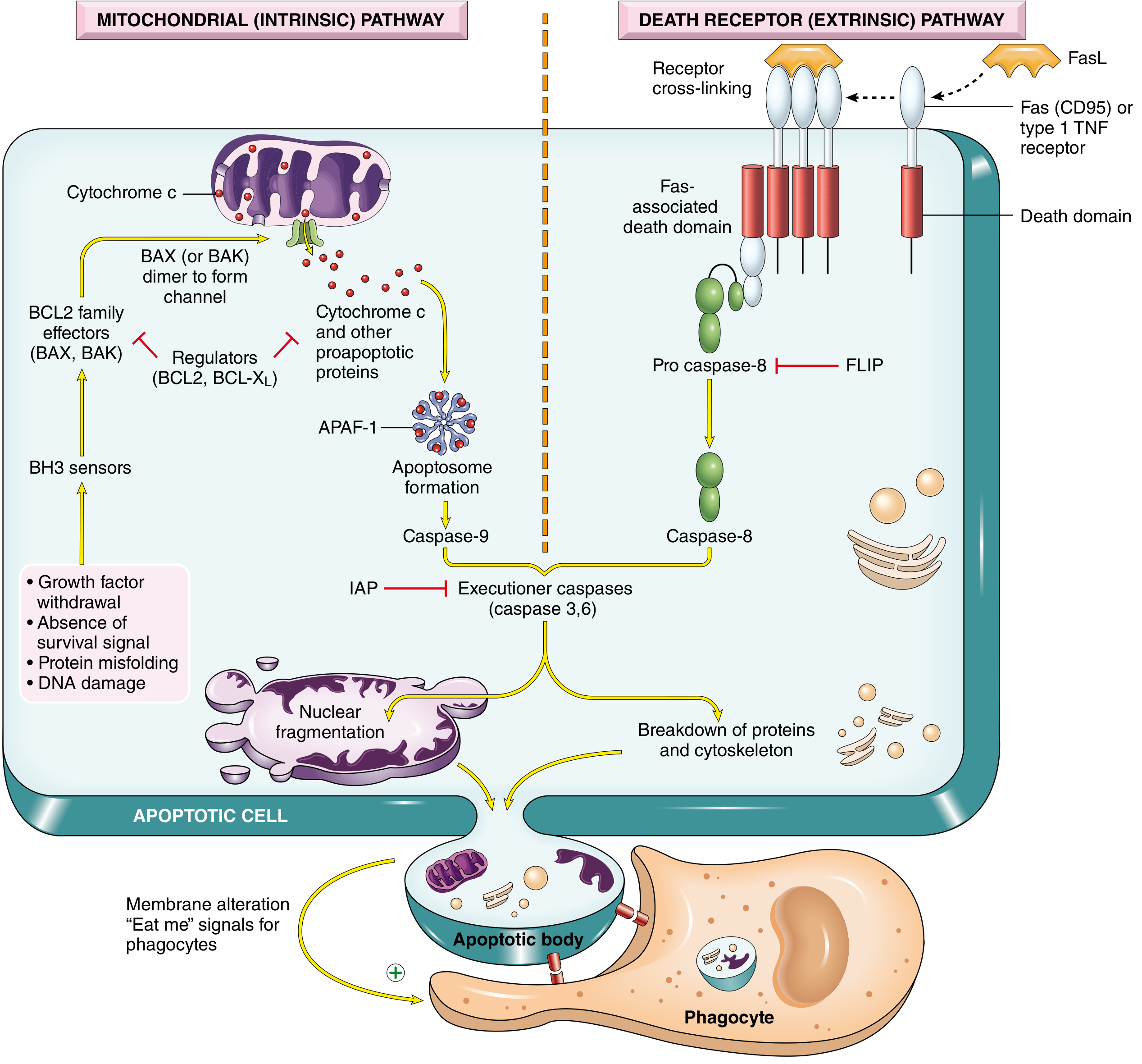
1. Mitochondrial (Intrinsic) Pathway
The predominant pathway in most physiologic and pathologic apoptosis.
Trigger signals:
- Loss of growth factors / survival signals
- DNA damage (e.g., radiation, chemotherapy)
- Oxidative stress, hypoxia
- Accumulation of misfolded proteins (ER stress)
Mechanism — step by step:
- Stress sensors activate BH3-only proteins (BAD, BIM, BID, PUMA, NOXA) — these are the molecular sensors of cellular damage.
- BH3-only proteins activate the pro-apoptotic effectors BAX and BAK, which oligomerize and insert into the outer mitochondrial membrane, forming pores.
- This increases mitochondrial outer membrane permeability → cytochrome c and other pro-apoptotic proteins (Smac/DIABLO) leak into the cytosol.
- Cytochrome c binds APAF-1 (apoptosis-activating factor-1) → forms the apoptosome (a heptameric wheel-shaped complex).
- The apoptosome recruits and activates caspase-9 (the initiator caspase).
- Active caspase-9 cleaves and activates executioner caspases-3 and -7 → cell death.
- Smac/DIABLO neutralizes IAPs (inhibitor of apoptosis proteins), removing the brake on caspase activation.
Regulators — the BCL-2 family:
| Group | Members | Action |
|---|---|---|
| Anti-apoptotic | BCL-2, BCL-XL, MCL-1 | Maintain outer mitochondrial membrane integrity; block cytochrome c release |
| Pro-apoptotic effectors | BAX, BAK | Permeabilize outer mitochondrial membrane |
| BH3-only sensors | BAD, BIM, BID, PUMA, NOXA | Activate BAX/BAK; neutralize BCL-2/BCL-XL |
The ratio of pro-apoptotic to anti-apoptotic BCL-2 family members determines whether a cell lives or dies. BCL-2 overexpression (seen in follicular B-cell lymphoma due to t(14;18)) blocks apoptosis and is a key oncogenic mechanism.
2. Death Receptor (Extrinsic) Pathway
Triggered by extracellular signals — important in immune regulation and cytotoxic T lymphocyte (CTL)-mediated killing.
Trigger signals:
- FasL (Fas ligand) binding Fas (CD95)
- TNF binding TNFR1
Mechanism — step by step:
- Death ligands (FasL, TNF) bind their death receptors (Fas/CD95, TNFR1) on the cell surface.
- Receptor trimerization → cytoplasmic death domains recruit the adaptor protein FADD (Fas-associated death domain).
- FADD recruits procaspase-8 → forms the DISC (death-inducing signaling complex).
- Procaspase-8 undergoes autocatalytic cleavage → active caspase-8.
- Caspase-8 directly activates executioner caspases-3 and -6 → cell death.
- In some cell types, caspase-8 also cleaves BID (a BH3-only protein) → truncated BID (tBID) enters mitochondria, amplifying the intrinsic pathway (cross-talk).
Inhibitor: FLIP (FLICE-like inhibitory protein) — binds procaspase-8 and blocks FADD binding, acting as a decoy that inhibits this pathway. Some viruses produce FLIP homologs to evade immune-mediated apoptosis.
Execution Phase
Both pathways converge on caspase-3, which:
- Activates a cytoplasmic DNase (CAD) that cleaves nuclear DNA into nucleosome-sized fragments (the "ladder" on gel electrophoresis)
- Degrades structural nuclear proteins (lamins) → nuclear collapse
- Breaks down cytoskeletal proteins → membrane blebbing
- Activates enzymes for final breakdown of cellular components
Physiologic vs. Pathologic Apoptosis
| Context | Examples |
|---|---|
| Physiologic | Embryonic development (tissue sculpting); elimination of self-reactive lymphocytes; post-immune response contraction; endometrial shedding |
| Pathologic | DNA damage beyond repair; viral infections; CTL killing of infected/tumor cells; duct obstruction (pancreas, kidney) |
Clinical Relevance
- Too little apoptosis → cancer (BCL-2 overexpression in follicular lymphoma), autoimmunity (failure to delete self-reactive T cells)
- Too much apoptosis → neurodegeneration (Alzheimer's, Parkinson's), ischemia-reperfusion injury, AIDS (CD4+ T cell depletion)
- Therapeutic targeting: BH3 mimetics (e.g., venetoclax/ABT-199, a BCL-2 inhibitor) are used clinically in CLL and AML to restore apoptosis in cancer cells
Sources: Robbins & Cotran Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Sleisenger & Fordtran's Gastrointestinal and Liver Disease
This is a shared conversation. Sign in to Orris to start your own chat.