How hypokalemia develops in conns syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I have everything needed for a thorough answer. Here is a detailed explanation:
How Hypokalemia Develops in Conn's Syndrome (Primary Hyperaldosteronism)
Conn's syndrome is caused by autonomous (non-renin-mediated) secretion of aldosterone by the adrenal cortex - most commonly from an aldosterone-producing adenoma (60-70% of cases) or bilateral adrenal hyperplasia. The excess aldosterone directly drives renal potassium wasting through a well-defined tubular mechanism.
Step 1: Aldosterone Acts on the Principal Cell of the Cortical Collecting Duct
Aldosterone is a mineralocorticoid that binds to the mineralocorticoid receptor (MR) in the principal cells of the cortical collecting duct (CCD) and connecting tubule (CNT). This is the primary site of regulated K⁺ excretion in the kidney.
Upon binding MR, aldosterone upregulates the expression and activity of three key proteins simultaneously:
| Protein | Location | Effect |
|---|---|---|
| ENaC (epithelial Na⁺ channel) | Apical membrane | More Na⁺ enters the cell from lumen |
| Na⁺/K⁺-ATPase | Basolateral membrane | Pumps Na⁺ out to blood, pulls K⁺ into cell |
| Apical K⁺ channels (ROMK) | Apical membrane | K⁺ secreted from cell into lumen |
- Comprehensive Clinical Nephrology, 7th Ed - "In the cortical collecting duct principal cell, K⁺ secretion involves integrated functions of the basolateral Na⁺, K⁺-ATPase, apical Na⁺ channel (ENaC), and apical K⁺ channel. Aldosterone stimulates this process through interaction with the mineralocorticoid receptor (MR), resulting in increased expression and activity of each of these processes."
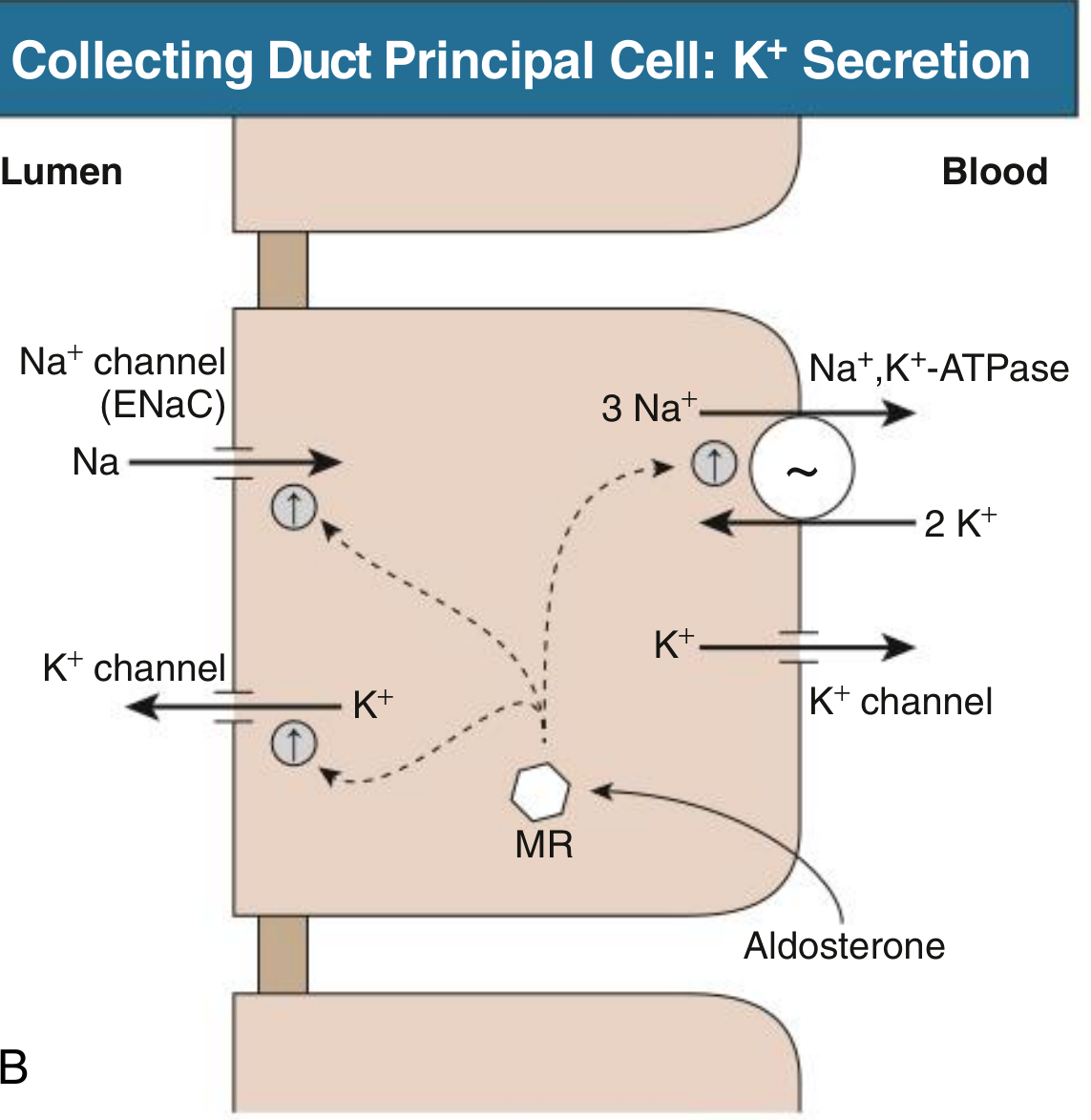
Step 2: The Electrochemical Driving Force for K⁺ Secretion
The key mechanism is electrogenic:
- Excess aldosterone causes massive Na⁺ reabsorption via ENaC from the tubular lumen into the principal cell
- This makes the tubular lumen electronegative relative to the cell interior (lumen-negative transepithelial potential difference)
- This electrical gradient acts as a powerful driving force pulling K⁺ (positively charged) out of the cell and into the lumen through apical K⁺ channels (ROMK)
- The basolateral Na⁺/K⁺-ATPase continuously replenishes intracellular K⁺ from the blood, keeping the cycle going
- K⁺ that accumulates in the lumen is then excreted in urine
In parallel, H⁺ is also secreted by alpha-intercalated cells (driven by the same lumen-negative potential), which explains the accompanying metabolic alkalosis.
- Harrison's Principles of Internal Medicine 22E - "Aldosterone activates the ENaC channel in principal cells via multiple synergistic mechanisms, thus increasing the driving force for K⁺ excretion."
Step 3: Volume Expansion Suppresses Renin - But K⁺ Loss Continues
Na⁺ retention causes mild volume expansion and raises blood pressure. This suppresses renin (and angiotensin II). However, the adenoma (or hyperplastic gland) continues secreting aldosterone autonomously, independent of renin - so the potassium-wasting drive is unrelenting.
- NKF Primer on Kidney Diseases, 8e - "Primary hyperaldosteronism is due to autonomous (non-renin-mediated) secretion of aldosterone by the adrenal cortex. This results in avid sodium retention and potassium secretion by the distal nephron. Patients present with volume-dependent hypertension, hypokalemia, and metabolic alkalosis."
Why Hypokalemia Occurs Despite Normal Dietary Intake
Normally, aldosterone secretion is feedback-inhibited by volume expansion and hyperkalemia would suppress it. In Conn's syndrome:
- The gland secretes aldosterone constitutively
- The kidney cannot compensate - it keeps losing K⁺ regardless of serum K⁺ levels
- Total body K⁺ depletion ensues
Clinical Note on Frequency
Importantly, frank hypokalemia (K⁺ < 3.5 mEq/L) is not universal - it occurs in fewer than 40% of patients with primary hyperaldosteronism, and is even less common with adrenal hyperplasia than with adenomas.
- Mulholland & Greenfield's Surgery, 7e - "Significant hypokalemia occurs in less than 40% of patients with primary hyperaldosteronism and is even less frequent in cases caused by adrenal hyperplasia, therefore hypokalemia is neither a sensitive nor specific marker of primary hyperaldosteronism."
Consequences of Hypokalemia in Conn's
| Effect | Mechanism |
|---|---|
| Muscle weakness and fatigue | K⁺ required for normal resting membrane potential |
| Polyuria / polydipsia | Hypokalemia impairs renal concentrating ability (nephrogenic DI) |
| Fasting hyperglycemia | K⁺ depletion impairs insulin secretion from beta cells |
| Metabolic alkalosis | Concurrent H⁺ loss from alpha-intercalated cells |
| ECG changes | Prolonged QT, U waves, ventricular arrhythmias |
Summary in One Sentence
Excess autonomous aldosterone in Conn's syndrome over-activates ENaC and Na⁺/K⁺-ATPase in collecting duct principal cells, creating a lumen-negative electrochemical gradient that continuously drives K⁺ into the urine, depleting total body potassium and producing hypokalemia.
This is a shared conversation. Sign in to Orris to start your own chat.