I now have everything needed to write a thorough, well-organized set of notes. Here are your comprehensive written notes on brain tumors:
BRAIN TUMORS - Comprehensive Written Notes
1. EPIDEMIOLOGY & OVERVIEW
- Annual incidence: ~23 per 100,000 persons for intracranial tumors; ~2 per 100,000 for intraspinal tumors
- CNS tumors account for ~20% of all childhood cancers - now the most deadly pediatric cancer (surpassing acute lymphoblastic leukemia)
- 70% of childhood CNS tumors arise in the posterior fossa (infratentorial); the majority of adult tumors arise supratentorially in the cerebral hemispheres
- Metastases are the most common tumors involving the CNS overall; meningiomas are the most common primary brain tumors
- Even "benign" or low-grade tumors can be lethal due to location (e.g., a benign posterior fossa meningioma compressing the medulla can cause cardiorespiratory arrest)
- Even the most malignant gliomas rarely metastasize outside the CNS; spread within CNS occurs via CSF (e.g., medulloblastoma "drop metastases")
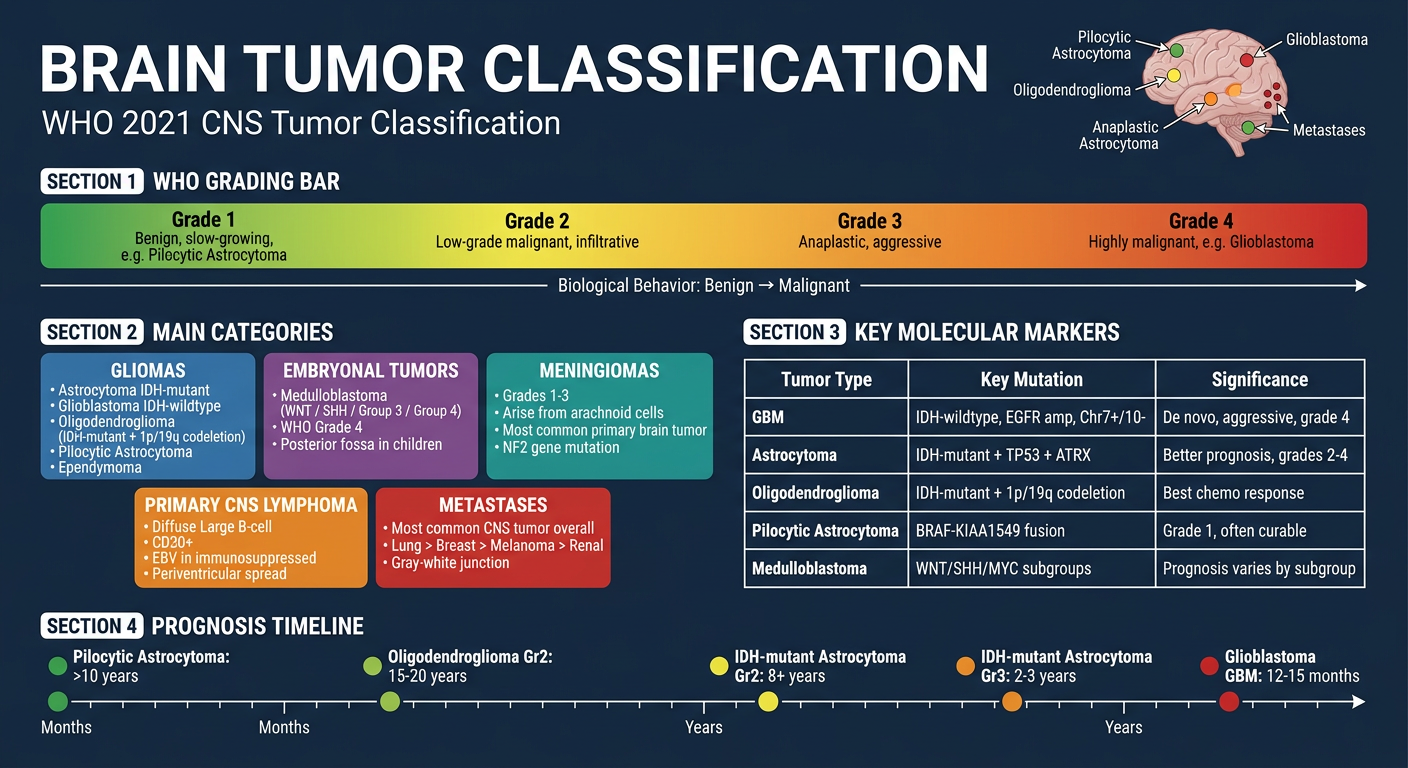
2. WHO 2021 CLASSIFICATION - FRAMEWORK
The 2021 WHO Classification of CNS Tumors integrates molecular/genetic markers with histology. Four grades are used:
| Grade | Behavior |
|---|
| CNS WHO Grade 1 | Benign, slow-growing (e.g., pilocytic astrocytoma) |
| CNS WHO Grade 2 | Low-grade malignant, infiltrative |
| CNS WHO Grade 3 | Anaplastic, more aggressive |
| CNS WHO Grade 4 | Highly malignant (e.g., GBM) |
Key molecular markers now required:
- IDH1/IDH2 mutation status - distinguishes astrocytoma (IDH-mutant) from glioblastoma (IDH-wildtype)
- 1p/19q codeletion - defines oligodendroglioma
- TERT promoter mutation - present in GBM and oligodendroglioma
- EGFR amplification, chr 7 gain / chr 10 loss - GBM signature
- ATRX, TP53 mutations - IDH-mutant astrocytoma
- CDKN2A/B deletion - upgrades IDH-mutant astrocytoma to Grade 4 even without histologic features
Major classes:
- Gliomas (astrocytomas, oligodendrogliomas, ependymomas)
- Neuronal/glioneuronal tumors
- Embryonal tumors (medulloblastoma)
- Meningiomas
- Primary CNS lymphoma
- Metastases
- Familial tumor syndromes (NF1, NF2, VHL, tuberous sclerosis)
3. PRIMARY TUMOR TYPES
3A. GLIOMAS
Gliomas are the most common group of primary intraparenchymal brain tumors. They arise from multipotent progenitor cells that differentiate down glial lineages.
3A-i. GLIOBLASTOMA (GBM) - IDH-wildtype, WHO Grade 4
The most aggressive and most common primary CNS malignancy.
- Accounts for ~14% of all primary CNS tumors and >50% of all CNS malignancies
- Age: predominantly >55 years, de novo (not from lower-grade precursors)
- Location: cerebral hemispheres
Molecular Pathogenesis:
| Alteration | Mechanism/Significance |
|---|
| IDH1/2 - wildtype | No IDH mutation; aggressive behavior |
| Chr 7 gain + Chr 10 loss | Most common combined alteration |
| TERT promoter mutation | Increased telomerase → senescence evasion |
| EGFR amplification | Activates growth factor signaling pathways |
| PDGFR amplification | Alternative growth signaling |
| CDKN2A deletion | Escape from normal growth controls |
| TP53 mutation | Resistance to apoptosis |
| PTEN loss | PI3K pathway activation |
Even an adult diffuse astrocytoma with IDH-wildtype and just ONE of the three key features (TERT mutation, EGFR amplification, or Chr 7+/10- changes) is classified as GBM regardless of histologic grade.
Morphology:
- Gross: Large, poorly defined infiltrative mass with central necrosis (required for diagnosis) and hemorrhage; typical "butterfly glioma" crossing corpus callosum
- Microscopy: Pseudopalisading necrosis (tumor cells line up around zones of necrosis), microvascular/endothelial proliferation, nuclear pleomorphism, brisk mitoses
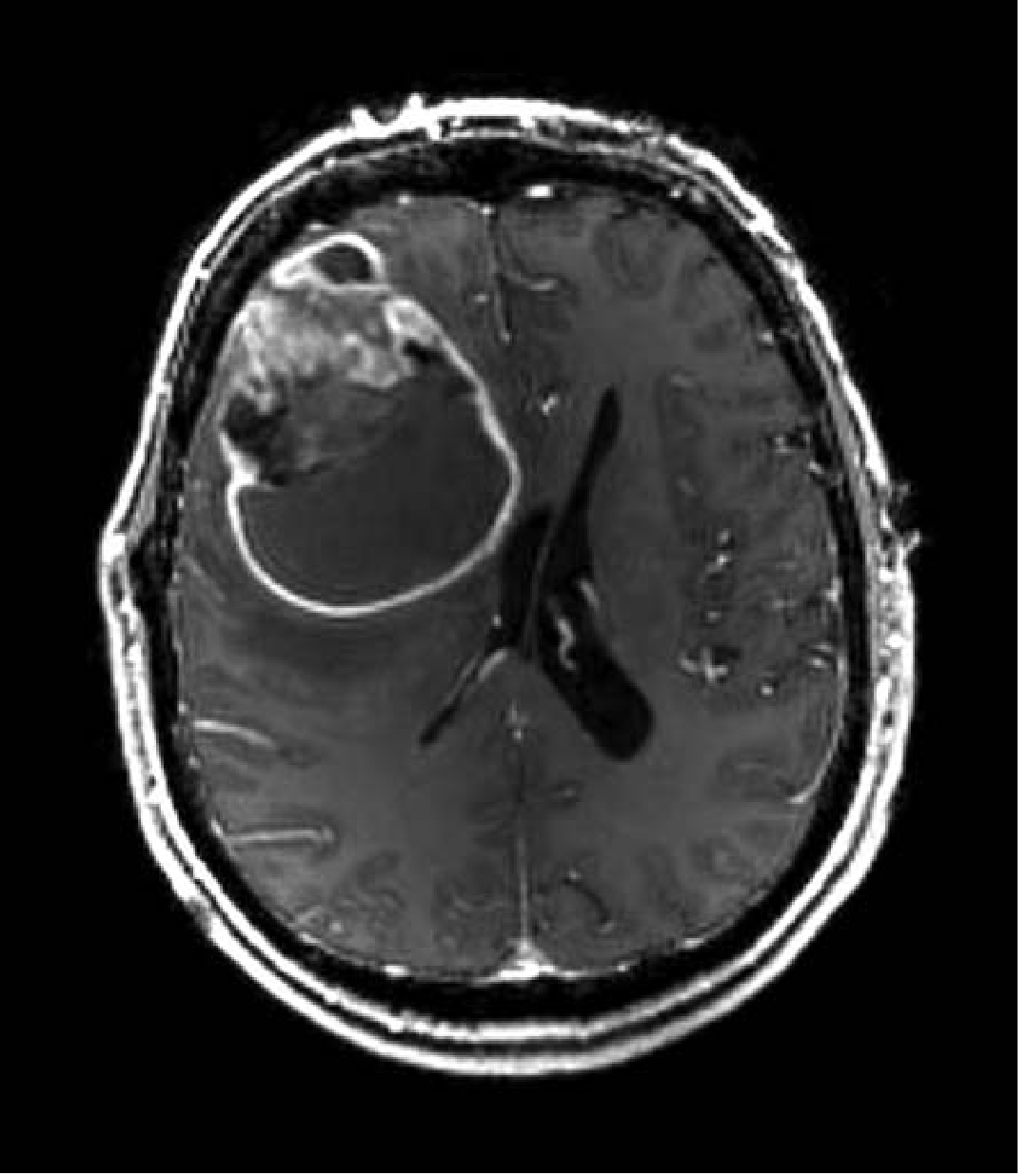
MRI appearance: Rim-enhancing mass on post-contrast T1; surrounding vasogenic edema on T2/FLAIR
MRI (post-contrast T1) of a Glioblastoma - rim-enhancing right frontal mass. - Robbins & Kumar Basic Pathology
Symptoms: Seizures, headaches, progressive focal neurologic deficits (depending on location), raised ICP signs (nausea/vomiting, papilledema), personality/cognitive changes. Rapid progression.
Prognosis: Median survival ~12-15 months with treatment; ~1 year overall.
3A-ii. ASTROCYTOMA, IDH-MUTANT (WHO Grades 2-4)
- More common in younger/middle-aged adults (median age 38 years)
- Location: cerebral hemispheres
- Significantly better prognosis than IDH-wildtype GBM, even at Grade 4
Molecular Pathogenesis:
- IDH1 or IDH2 gain-of-function mutation → production of 2-hydroxyglutarate (oncometabolite) → inhibits alpha-ketoglutarate-dependent dioxygenases → hypermethylation of DNA and histones → epigenetic silencing of tumor suppressor genes
- Co-occurring mutations: TP53 (apoptosis resistance) and ATRX inactivation (alternative lengthening of telomeres)
- Grade upgrade to 4: homozygous CDKN2A/B deletion
Grading within IDH-mutant astrocytoma:
| Grade | Features |
|---|
| 2 | Mild-moderate hypercellularity, nuclear pleomorphism, GFAP-positive; no mitoses/necrosis/microvascular proliferation |
| 3 | Increased mitotic activity + nuclear atypia |
| 4 | Necrosis OR microvascular proliferation OR homozygous CDKN2A/B deletion |
Morphology:
- Grade 2-3: Poorly defined, gray, infiltrative; expand brain without forming discrete mass
- Grade 4 (IDH-mutant): diffuse infiltration but usually lacks large central necrosis/hemorrhage seen in GBM
- GFAP-positive on immunohistochemistry
Prognosis: Median survival 8+ years (Grade 2), 2-3 years (Grade 3), better than GBM even at Grade 4
3A-iii. OLIGODENDROGLIOMA, IDH-Mutant + 1p/19q Codeleted (WHO Grades 2-3)
- 5-15% of gliomas; peak 4th-5th decade
- Location: cerebral hemispheres (frontal/temporal lobes predominantly)
- Best prognosis among diffuse glial tumors
Molecular Pathogenesis:
- Codeletion of chromosomes 1p and 19q - always in association with IDH1/IDH2 mutation
- Most also have TERT promoter mutation (increased telomerase activity)
- The 1p/19q codeletion is a marker of chemosensitivity
Morphology:
- Gross: Gelatinous, gray masses; cysts, focal hemorrhage, calcification (in up to 90%)
- Microscopy: Sheets of regular cells with spherical nuclei + clear perinuclear halo ("fried egg" appearance); delicate "chicken wire" vascular pattern (anastomosing capillaries)
- Perineuronal satellitosis when infiltrating cortex
Symptoms: Seizures (often years before diagnosis), headaches, focal neurologic deficits
Prognosis:
- Grade 2: Average survival 10-20 years with surgery + chemo + RT
- Grade 3: 5-10 years
3A-iv. PILOCYTIC ASTROCYTOMA (WHO Grade 1)
- Most common pediatric brain tumor (children and young adults)
- Location: cerebellum (most common), 3rd ventricle region, optic nerves/chiasm, spinal cord
- Benign, circumscribed - NOT infiltrative; often curable with resection alone
Molecular Pathogenesis:
- KIAA1549-BRAF gene fusion/duplication in 80-90% (especially cerebellar tumors) - activates MAPK/ERK pathway; associated with better overall survival
- NOT IDH-mutant; distinct from diffuse gliomas
Morphology:
- Gross: Well-demarcated, often cystic with a contrast-enhancing mural nodule
- Microscopy: Biphasic architecture - loose "microcystic" areas + compact fibrillar areas; Rosenthal fibers (eosinophilic corkscrew-shaped inclusions) + eosinophilic granular bodies (mulberry-like); GFAP-positive bipolar "hairlike" processes
- Unlike diffuse gliomas: microvascular proliferation or necrosis does NOT imply bad prognosis here
Symptoms: Signs of posterior fossa mass effect - ataxia, obstructive hydrocephalus, raised ICP (headache, vomiting)
Prognosis: >90% 5-year survival after surgery; recurrence risk if brainstem involvement. Incompletely resected lesions treated with chemo or targeted therapy (BRAF inhibitors).
3A-v. EPENDYMOMA (WHO Grades 2-3)
- Arise near the ependymal-lined ventricular system and the central canal of the spinal cord
- In children (1st-2nd decade): near the 4th ventricle (5-10% of pediatric brain tumors); in adults: spinal cord more common
- Location strongly influences prognosis
Molecular Pathogenesis (key gene fusions):
- RELA fusion (supratentorial ependymoma, more aggressive)
- YAP1 fusion
- Posterior fossa group A (PF-A): H3K27me3 loss, worse prognosis; occurs in young children
- Posterior fossa group B (PF-B): better prognosis, older patients
Morphology:
- Gross: Well-defined, solid or partially cystic
- Microscopy: Perivascular pseudorosettes (tumor cells oriented around blood vessels with anuclear zones of cytoplasmic processes) and true ependymal rosettes (cylindrical arrangements around a central lumen)
- GFAP-positive
Symptoms: 4th ventricle tumors: hydrocephalus (headache, vomiting, papilledema), ataxia, cranial nerve palsies. Spinal: back pain, limb weakness, bladder/bowel dysfunction.
3B. MEDULLOBLASTOMA (WHO Grade 4, Embryonal Tumor)
- Most common malignant pediatric brain tumor; highly aggressive embryonal tumor
- Arises in the cerebellar vermis in children (posterior fossa); lateral cerebellum in adults
- Accounts for up to 20% of pediatric CNS tumors
Molecular Subgroups (4 main groups with major prognostic differences):
| Subgroup | Key Feature | Prognosis |
|---|
| WNT-activated | CTNNB1 mutation (beta-catenin), classic histology | ~100% 5-year survival (best) |
| SHH-activated | PTCH1/SUFU/SMO mutation; desmoplastic/nodular histology | Variable (PTCH1 adult - good; TP53-mutant - poor) |
| Group 3 | MYC amplification, large cell/anaplastic | 20-30% 5-year survival (worst) |
| Group 4 | CDK6 amp, i17q; most common (35%) | Intermediate |
Morphology:
- Gross: Gray, friable mass often in cerebellar vermis; may extend to leptomeninges
- Microscopy: Densely cellular sheets of small, primitive cells with scant cytoplasm, hyperchromatic/crescent-shaped nuclei; abundant mitoses; Homer Wright rosettes (neuronal differentiation; also in neuroblastoma); synaptophysin-positive; GFAP less common
- Desmoplastic subtype: collagen/reticulin deposition + "pale islands" (nodules of neuropil)
- Large cell/anaplastic: large vesicular nuclei, prominent nucleoli, cell wrapping, high mitotic/apoptotic indices
CSF dissemination: Tumor spreads through CSF → "icing" on brain surface OR nodular "drop metastases" at cauda equina (very characteristic)
Symptoms: Hydrocephalus (headache, vomiting), ataxia, truncal instability, morning vomiting (classic in children), diplopia, cranial nerve palsies
Prognosis: Highly sensitive to radiation and chemotherapy. WNT subtype ~100% survival; aggressive subtypes 20-30%.
3C. MENINGIOMA (WHO Grades 1-3)
- Most common primary brain tumor overall
- Arise from meningothelial (arachnoid cap) cells of the arachnoid; attached to dura
- Adults (usually >40), female predominance (2:1)
- May occur along any external brain surface, within ventricles, or at skull base
Molecular Pathogenesis:
- ~50% sporadic meningiomas: somatic loss-of-function mutations in NF2 tumor suppressor gene (found in all grades - involved in tumor initiation)
- Multiple meningiomas + 8th nerve schwannomas → think Neurofibromatosis Type 2 (NF2)
- Non-NF2 sporadic tumors: mutations in Hedgehog pathway regulators, TRAF7, AKT1, SMO, KLF4
- Grade 3 markers: BAP1 mutation, TERT promoter mutation, homozygous CDKN2A/B deletion
WHO Grading:
| Grade | Histology | Behavior |
|---|
| 1 (Benign) | Meningothelial, fibrous, transitional, psammomatous, angiomatous, secretory | 80-90% of cases; low recurrence |
| 2 (Atypical) | Increased mitoses, sheeting, small cells, prominent nucleoli, necrosis, brain invasion | Higher recurrence |
| 3 (Anaplastic/Malignant) | Overtly malignant cytology, high mitoses | Aggressive; rare |
Psammoma bodies: Concentric calcified spherules - characteristic of psammomatous meningioma
Morphology:
- Gross: Well-circumscribed, firm, bosselated (lobulated), rubbery mass attached to dura; usually easily separated from brain (except infiltrative variants)
- Microscopy: Whorled pattern of meningothelial cells; psammoma bodies; no necrosis (Grade 1)
Symptoms: Often vague, non-localizing initially. Focal symptoms depend on location:
- Parasagittal: contralateral leg weakness/sensory loss
- Sphenoid ridge: facial pain, visual loss, proptosis
- Olfactory groove: anosmia, frontal lobe signs
- Posterior fossa: SNHL (sensorineural hearing loss), tinnitus, imbalance, facial paresthesia, diplopia, ataxia
- Seizures (from cortical compression), raised ICP signs
Prognosis: Grade 1 - very good after gross total resection; Grade 2 and 3 - higher recurrence and need adjuvant radiation.
3D. PRIMARY CNS LYMPHOMA (PCNSL)
- 2% of extranodal lymphomas; 1% of intracranial tumors
- Most common CNS neoplasm in immunosuppressed individuals (AIDS, post-transplant)
- In immunocompetent: frequency increases after age 60
- Histology: almost always diffuse large B-cell lymphoma (aggressive); worse outcomes than same histology at non-CNS sites
- In immunosuppressed: malignant B cells are latently infected with Epstein-Barr virus (EBV)
Morphology:
- Often multifocal; involves deep gray structures, white matter, cortex; periventricular spread common
- Better defined than gliomas but less discrete than metastases
- EBV-associated: extensive necrosis
- Microscopy: Malignant lymphoid cells accumulate perivascularly and infiltrate brain parenchyma
- IHC: CD20 positive (B-cell marker)
Symptoms: Focal neurologic deficits, headache, cognitive/behavioral changes, uveitis (may involve the eye), seizures (less common)
3E. BRAIN METASTASES
- Most common tumors involving the CNS overall (more common than primary tumors)
- Common primaries: lung (most common), breast, melanoma, renal cell, colon
- Typically well-demarcated, often at the gray-white junction (where blood flow slows)
- Often multiple; surrounded by disproportionate vasogenic edema
- Microscopy: Tumor emboli, necrosis; histology reflects the primary site
Mechanism: Hematogenous spread → arrest at gray-white junction → breach of blood-brain barrier → tumor seeding. Surrounding edema from leaky vasculature (VEGF-mediated)
Symptoms: Focal neurologic deficits, seizures, headache (raised ICP), behavioral/cognitive changes
4. CLINICAL FEATURES & SYMPTOMS (All Brain Tumors)
General/Non-Focal Symptoms (Raised ICP)
| Symptom | Mechanism |
|---|
| Headache | CSF flow obstruction + raised ICP; worse with Valsalva, bending over, waking from sleep; initially dull/morning headache |
| Nausea/Vomiting | Raised ICP (especially in adults with malignant tumors) |
| Papilledema | Raised ICP transmitted to optic nerve sheath |
| Altered consciousness/lethargy | Diffuse ICP effect on reticular activating system |
| Herniation syndromes | Uncal herniation: ipsilateral CN III palsy (blown pupil) + contralateral hemiplegia; Tonsillar herniation: sudden death |
Focal Symptoms (Localizing)
| Location | Symptoms |
|---|
| Frontal lobe | Personality change, disinhibition, impaired executive function, contralateral hemiparesis (motor cortex), expressive aphasia (Broca's area - dominant) |
| Temporal lobe | Receptive aphasia (Wernicke's - dominant), complex partial seizures, memory deficits, contralateral visual field defect (superior quadrantanopia) |
| Parietal lobe | Contralateral sensory loss, neglect (non-dominant), Gerstmann syndrome (dominant: acalculia, agraphia, finger agnosia, L-R disorientation) |
| Occipital lobe | Contralateral homonymous hemianopia, visual agnosia |
| Cerebellum | Ipsilateral ataxia, dysmetria, dysdiadochokinesia, intention tremor, truncal instability |
| Brainstem | Cranial nerve palsies, long-tract signs (hemi/quadriplegia), diplopia, dysphagia |
| Corpus callosum | Disconnection syndromes, "butterfly" GBM |
| Thalamus/Basal ganglia | Contralateral sensory loss, movement disorders |
| Pineal region | Parinaud syndrome (vertical gaze palsy, light-near dissociation, convergence-retraction nystagmus) + hydrocephalus |
Seizures
- Due to cortical irritation by the tumor or surrounding edema
- New onset seizures in adults: always suspect brain tumor until proven otherwise
- More common with slow-growing tumors (e.g., oligodendroglioma, low-grade astrocytoma)
Headache Red Flags Suggesting Brain Tumor
- Worsened by Valsalva maneuver
- Waking patient from sleep
- Abnormal neurologic exam
- Progressive worsening
- New-onset in patient >50 years
- Associated with seizures or mental status change
5. DIAGNOSIS & INVESTIGATIONS
Imaging
- MRI with and without gadolinium: gold standard
- Best for soft-tissue detail, posterior fossa, brainstem
- Post-contrast T1: enhancement pattern (ring = GBM/metastasis; homogeneous = low-grade glioma/meningioma)
- T2/FLAIR: edema extent, non-enhancing tumor infiltration
- DWI: restriction in highly cellular tumors and CNS lymphoma
- MR Spectroscopy: ↑Cho/NAA ratio in tumors; ↑lactate in necrotic regions
- Perfusion MRI (rCBV): distinguish high vs. low grade; pseudoprogression vs. true progression
- fMRI + DTI: mapping eloquent cortex and white matter tracts before surgery
- CT scan:
- Faster; good for emergencies; can detect hemorrhage (meningioma + oligodendroglioma calcifications well seen)
- Noncontrast CT: identifies large masses, edema, hemorrhage, calcification
Tissue Diagnosis
- Stereotactic needle biopsy: for lesions in eloquent/deep areas
- Surgical resection: diagnostic + therapeutic (gross total resection preferred)
- Molecular testing: IDH1/2, 1p/19q, MGMT promoter methylation, EGFR, TERT, ATRX, Ki-67
MGMT Methylation Status
- MGMT (O6-methylguanine-DNA methyltransferase): DNA repair enzyme that reverses alkylation damage from chemotherapy
- MGMT promoter methylation → silenced MGMT → tumor cells cannot repair temozolomide-induced DNA damage → better response to temozolomide
- Present in ~40% of GBMs; strong predictor of improved survival with temozolomide
Other Investigations
- EEG: for seizure evaluation
- Lumbar puncture: CSF cytology for leptomeningeal disease (caution: contraindicated with raised ICP)
- Ophthalmology: assess optic discs (papilledema), PCNSL eye involvement
6. MANAGEMENT
Surgical Principles
- Gross total resection (GTR) is the goal whenever safe - improves survival for all grades
- Eloquent area tumors (motor cortex, language centers, brainstem, deep midline structures): may limit extent of resection → biopsy or subtotal resection
- Advanced techniques: awake craniotomy with brain mapping (language/motor mapping), intraoperative MRI, 5-ALA fluorescence-guided resection (GBM), diffusion tensor imaging (DTI) to preserve white matter tracts, functional MRI (fMRI)
- Stereotactic needle biopsy: used for deep/eloquent lesions not amenable to open surgery
Treatment by Tumor Type
GBM (IDH-wildtype, Grade 4)
- Maximal safe surgical resection (gross total or near-total)
- Stupp Protocol (standard of care):
- Concurrent radiotherapy (60 Gy in 30 fractions over 6 weeks) + temozolomide (TMZ) (75 mg/m²/day during RT)
- Then adjuvant TMZ (150-200 mg/m² days 1-5, every 28 days x 6 cycles)
- Median survival improved from ~12 months (RT alone) to ~14-16 months with TMZ
- MGMT methylated GBM: much better response to TMZ; median survival 21+ months
- Bevacizumab (anti-VEGF antibody): Used for recurrent GBM; reduces edema + delays progression; does not improve overall survival convincingly
- Tumor Treating Fields (Optune): Electric field device worn on scalp; disrupts mitosis; added to TMZ in newly diagnosed GBM → modest survival benefit
- Dexamethasone: For cerebral edema management (reduces vasogenic edema around tumor)
- Antiepileptic drugs (AEDs) for seizure management
IDH-Mutant Astrocytoma (Grades 2-4)
- Surgery (maximal resection) + radiation therapy (conformal RT, ~54-60 Gy)
- Chemotherapy: Temozolomide (first-line) OR PCV (procarbazine, CCNU/lomustine, vincristine) - PCV in addition to RT showed 13.8 vs 7.8 year median survival
- Grade 2: In younger patients with normal neuro exam, radiation may be delayed; serial imaging surveillance is acceptable
- Grade 3-4: radiation + chemotherapy concurrently
- Seizure management: AEDs (levetiracetam preferred - no drug interactions with chemotherapy)
Oligodendroglioma (IDH-mutant + 1p/19q codeleted, Grades 2-3)
- Most chemosensitive glioma due to 1p/19q codeletion
- Surgery + RT + chemotherapy (PCV or TMZ)
- Average survivals: 15-20 years (Grade 2); 10-15 years (Grade 3)
Pilocytic Astrocytoma (Grade 1)
- Complete surgical resection = curative (cerebellar location is ideal)
- 5-year survival >90% after successful surgery
- Recurrence or incomplete resection: repeat surgery, chemotherapy (carboplatin + vincristine in children), or BRAF inhibitors (dabrafenib + trametinib for BRAF-fusion positive)
- Radiation for unresectable tumors
Medulloblastoma
- Exquisitely sensitive to radiation and chemotherapy
- Standard: surgery (maximal resection of cerebellar tumor) + craniospinal irradiation (CSI) + chemotherapy (cisplatin, vincristine, lomustine/CCNU)
- Risk-stratified treatment:
- Average risk: 23.4 Gy CSI + posterior fossa boost + chemotherapy
- High risk (M+ disease, residual tumor >1.5 cm²): 36 Gy CSI + boost + chemo
- WNT-activated: clinical trials exploring reduced-intensity treatment (excellent prognosis)
- SHH-activated: SMO inhibitors (vismodegib, sonidegib) in trials
- Prognosis: WNT ~100%; Group 3 (MYC-amp) 20-30%
Meningioma
- Grade 1, asymptomatic/small: surveillance with serial MRI (many grow slowly or not at all)
- Symptomatic or growing Grade 1: surgical resection (GTR) → low recurrence
- Grade 2: GTR + adjuvant RT (stereotactic radiosurgery or fractionated RT)
- Grade 3: aggressive surgery + RT; systemic therapy for refractory disease (hydroxyurea, somatostatin analogs for some)
- Radiosurgery (Gamma Knife/CyberKnife): excellent for small, deep, or surgically inaccessible meningiomas; local control rates >90%
Primary CNS Lymphoma
- NOT treated with surgery (biopsy only for diagnosis - avoid corticosteroids before biopsy as they lyse lymphoma cells and reduce diagnostic yield)
- High-dose methotrexate (HD-MTX) based regimens: backbone of treatment (MTX crosses BBB at high doses)
- Consolidation: whole brain RT (higher neurotoxicity) or autologous stem cell transplant in younger patients
- Rituximab (anti-CD20): added to MTX regimens
- In AIDS: treat HIV first (HAART); EBV-driven tumors may regress with immune reconstitution
- Prognosis: 2-year survival ~60% in immunocompetent with HD-MTX; dismal in untreated patients
Brain Metastases
- Single accessible metastasis: surgical resection + whole brain RT (WBRT) → improves median survival 40 weeks; OR stereotactic radiosurgery (SRS)
- Multiple metastases: WBRT; SRS (Gamma Knife) can be applied to multiple metastases in one session with improved outcomes
- Systemic therapy: targeted agents if known driver mutation (e.g., erlotinib for EGFR-mutant NSCLC, lapatinib for HER2+ breast; some cross the BBB)
- Dexamethasone: mainstay for controlling cerebral edema and symptoms
- Immunotherapy (checkpoint inhibitors): emerging role in melanoma, NSCLC brain metastases
Supportive/Symptomatic Management
| Symptom/Problem | Management |
|---|
| Cerebral edema / raised ICP | Dexamethasone (4-16 mg/day in divided doses); osmotherapy (mannitol, hypertonic saline) for acute ICP crises |
| Seizures | Levetiracetam (preferred), valproate; prophylactic AEDs controversial in non-seizing patients |
| Hydrocephalus (obstructive) | Ventriculoperitoneal (VP) shunt; endoscopic 3rd ventriculostomy; external ventricular drain (EVD) acutely |
| Venous thromboembolism | High risk in brain tumor patients; low-molecular-weight heparin (LMWH) if no hemorrhage risk |
| Fatigue/depression | Methylphenidate for cancer-related fatigue; antidepressants |
| Cognitive decline | Memantine (reduces cognitive effects of WBRT); cognitive rehabilitation |
| Nausea/vomiting | Ondansetron, metoclopramide; dexamethasone |
7. MOLECULAR MECHANISM SUMMARY TABLE
| Tumor | Key Molecular Mechanism | Oncometabolite / Driver |
|---|
| GBM (IDH-WT) | Chr 7+/10-, TERT mutation, EGFR amplification, PTEN loss → uncontrolled proliferation + evasion of senescence | None (no oncometabolite) |
| Astrocytoma (IDH-mutant) | IDH1/2 gain-of-function → 2-hydroxyglutarate → DNA/histone hypermethylation → tumor suppressor silencing + TP53 + ATRX mutations | 2-HG (oncometabolite) |
| Oligodendroglioma | IDH1/2 mutation + 1p/19q codeletion (loss of tumor suppressors on chr 1p: FUBP1; chr 19q: CIC) + TERT mutation | 2-HG |
| Pilocytic Astrocytoma | KIAA1549-BRAF fusion → constitutive MAPK/ERK activation → proliferation | BRAF fusion protein |
| Medulloblastoma WNT | CTNNB1 mutation → beta-catenin nuclear accumulation → WNT target gene activation | WNT pathway |
| Medulloblastoma SHH | PTCH1/SUFU loss → constitutive SMO/GLI activation → proliferation | SHH pathway |
| Medulloblastoma Gr 3 | MYC amplification → global transcription → rapid proliferation | MYC |
| Meningioma | NF2 loss (merlin) → uncontrolled contact inhibition failure; AKT1/SMO mutations → PI3K/mTOR/Hedgehog pathway | NF2/merlin loss |
| PCNSL | EBV LMP1 mimics CD40 → constitutive NF-kB → B-cell proliferation (in immunosuppressed); MYD88 L265P mutation (in immunocompetent) | EBV / MYD88 |
| GBM angiogenesis | HIF-1α → VEGF overexpression → tumor neovascularity → leaky BBB → edema | VEGF |
8. FAMILIAL/HEREDITARY SYNDROMES
| Syndrome | Gene | CNS Tumors |
|---|
| Neurofibromatosis Type 1 (NF1) | NF1 (neurofibromin; 17q11) | Optic gliomas (pilocytic astrocytoma), brainstem gliomas, spinal neurofibromas |
| Neurofibromatosis Type 2 (NF2) | NF2 (merlin; 22q12) | Bilateral vestibular schwannomas (pathognomonic), meningiomas, ependymomas |
| Von Hippel-Lindau (VHL) | VHL (3p25) | Cerebellar/retinal hemangioblastomas |
| Tuberous Sclerosis | TSC1 (hamartin), TSC2 (tuberin) | Cortical tubers, subependymal giant cell astrocytoma (SEGA) |
| Li-Fraumeni Syndrome | TP53 | GBM, other brain tumors |
| Gorlin Syndrome (Basal Cell Nevus) | PTCH1 | Medulloblastoma (desmoplastic/SHH subtype) |
| Cowden Syndrome | PTEN | LDD (Lhermitte-Duclos disease - cerebellar gangliocytoma) |
9. QUICK COMPARISON TABLE
| Feature | GBM | IDH-mutant Astrocytoma | Oligodendroglioma | Meningioma | Medulloblastoma |
|---|
| Age | >55 yrs | 30-40 yrs | 40-50 yrs | >40 yrs (F>M) | Children |
| Location | Cerebral hemispheres | Cerebral hemispheres | Frontal/temporal | Dural surface | Cerebellar vermis |
| WHO Grade | 4 | 2-4 | 2-3 | 1-3 | 4 |
| Key molecular | IDH-WT, EGFR amp, chr 7+/10- | IDH-mutant, TP53, ATRX | IDH-mutant, 1p/19q codeletion | NF2 loss | WNT/SHH/MYC/CDK6 |
| Histology clue | Pseudopalisading necrosis | GFAP+ infiltrative | "Fried egg" + calcification | Whorls + psammoma bodies | Homer Wright rosettes |
| Prognosis | 12-15 months | Years | 10-20 years | Mostly curable | Subtype-dependent |
| Key treatment | Surgery + RT + TMZ (Stupp) | Surgery + RT + PCV/TMZ | Surgery + RT + PCV/TMZ | Surgery ± RT | Surgery + CSI + chemo |
SOURCES
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th Ed. - CNS Tumors chapter
- Robbins & Kumar Basic Pathology - CNS Tumors chapter
- Adams and Victor's Principles of Neurology, 12th Ed. - Glioma/Astrocytoma sections
- Schwartz's Principles of Surgery, 11th Ed. - Glial Tumors chapter
- Tintinalli's Emergency Medicine: A Comprehensive Study - Brain Tumor section