Pathogenesis of basal cell carcinoma
"basal cell carcinoma"[MeSH Terms] AND pathogenesis
Pathogenesis of Basal Cell Carcinoma (BCC)
1. Cell of Origin
- Fitzpatrick's Dermatology, Vol. 1-2 (Fitzpatrick's), p. 1913
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1059
2. The Central Molecular Hallmark: Hedgehog Pathway Activation
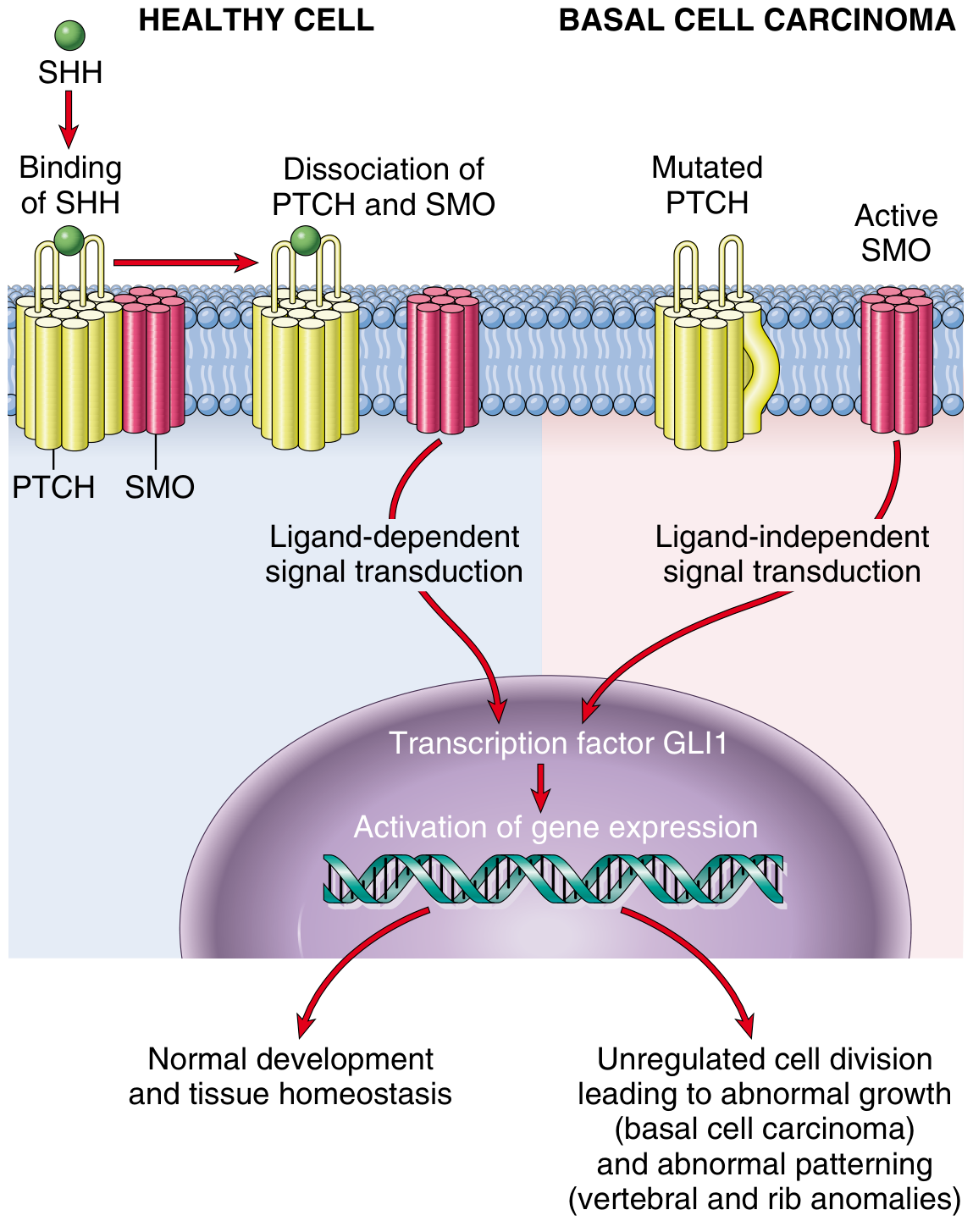
Normal Hedgehog Signaling
- PTCH1 (Patched-1), the receptor for SHH, forms a complex with SMO (Smoothened) and sequesters it in an inactive state outside the primary cilium.
- PTCH1 inhibits SMO activity.
- GLI transcription factors (GLI1, GLI2, GLI3) are suppressed by SUFU (Suppressor of Fused).
- No proliferative genes are transcribed.
- SHH binds PTCH1 → releases SMO into the primary cilium → SMO inhibits SUFU → GLI transcription factors activate → expression of proliferative and survival genes.
Oncogenic Activation in BCC
| Gene | Type of Mutation | Prevalence in Sporadic BCC |
|---|---|---|
| PTCH1 | Loss-of-function (tumor suppressor) | ~73% |
| SMO | Gain-of-function (oncogene) | ~20% |
| SUFU | Loss-of-function | ~8% |
- ~90% of sporadic BCCs have identifiable mutations in at least one PTCH1 allele.
- An additional ~10% have activating mutations in SMO.
- In both cases, the result is constitutive activation of GLI1, driving unregulated transcription of genes promoting cell growth, survival, and proliferation.
3. Role of UV Radiation
- UVB directly damages DNA, causing characteristic C→T and CC→TT transition mutations at dipyrimidine sites (the "UV signature").
- Approximately one-third of PTCH1 mutations in sporadic BCC are C→T transitions, the hallmark of UV damage.
- TP53 mutations (also UV-induced) are found in ~50-61% of BCC cases. TP53 normally triggers cell cycle arrest and apoptosis in response to DNA damage; its loss allows mutated cells to survive and proliferate.
- A latency period of 20-50 years typically separates the time of UV damage from clinical onset, explaining why BCC predominantly presents in older adults.
- Incidence is 40-fold higher in sunny equatorial climates (e.g., Australia) compared to Northern European locales.
4. Germline Mutations: Nevoid BCC Syndrome (Gorlin Syndrome / BCNS)
- Patients develop hundreds of BCCs from a young age.
- The syndrome also includes odontogenic keratocystic tumors, medulloblastoma, bifid ribs, calcification of the falx cerebri, and subtle developmental anomalies (reflecting HH's role in embryonic patterning).
- A second somatic hit (usually UV-induced) inactivates the remaining PTCH1 allele, following the two-hit tumor suppressor model.
- Importantly, radiation therapy is contraindicated in BCNS patients because ionizing radiation dramatically accelerates BCC development.
5. Additional Pathogenic Factors
| Factor | Mechanism |
|---|---|
| TP53 mutations | UV-induced; loss of apoptosis checkpoint (~50-61% of BCCs) |
| Ionizing radiation | Direct DNA damage; can cause BCC at non-sun-exposed sites |
| Immunosuppression | Impaired immunosurveillance (organ transplant recipients have markedly elevated risk) |
| Xeroderma pigmentosum | Nucleotide excision repair deficiency → UV damage accumulates unrepaired |
| Arsenic exposure | Causes BCC at sun-protected sites; mechanism involves DNA damage + impaired repair |
| GWAS findings | 14+ SNPs identified involving telomere maintenance, immune regulation, and tumor progression |
6. Morphological Consequences of Pathogenesis
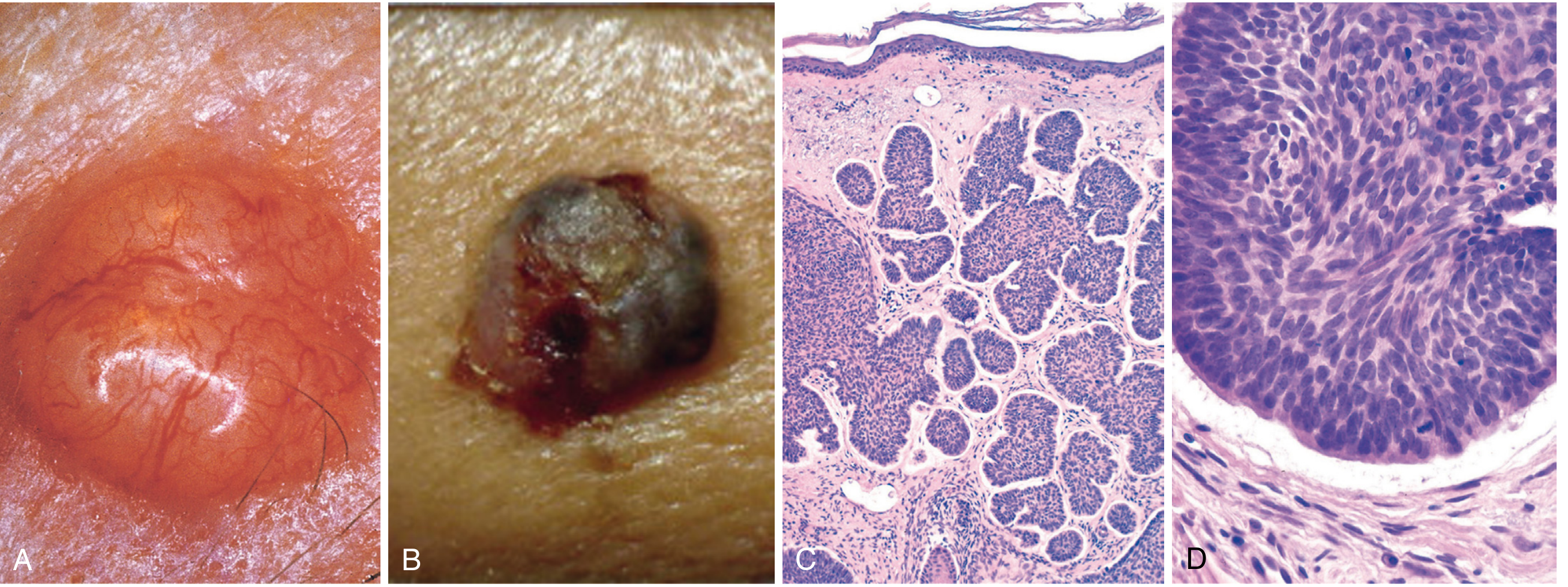
- Nests and islands of basaloid cells with peripheral nuclear palisading
- Characteristic peritumoral clefts (stromal retraction from tumor islands)
- Fibrotic or mucinous stromal matrix
- Cells arise only from epidermis or follicular epithelium (never mucosa)
- Tumor cells closely resemble normal basal cells with large nuclei and scant cytoplasm; mitoses are usually absent or infrequent
7. Therapeutic Relevance of Pathogenesis
- Vismodegib and sonidegib - small-molecule SMO inhibitors - are approved for locally advanced or metastatic BCC that cannot be treated surgically.
- Immune checkpoint inhibitors (e.g., cemiplimab) are used for advanced disease.
Summary
| Step | Event |
|---|---|
| 1 | Chronic UVB exposure → C→T mutations in PTCH1, TP53 |
| 2 | PTCH1 loss-of-function → SMO constitutively active |
| 3 | Active SMO → SUFU inhibited → GLI1/2/3 nuclear entry |
| 4 | GLI1 transcription → unregulated basaloid cell proliferation |
| 5 | TP53 mutations → impaired apoptosis checkpoint |
| 6 | Additional hits (immunosuppression, ionizing radiation) amplify risk |
| 7 | Clinically: slow-growing, locally invasive, rarely metastatic tumor |
- Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.), pp. 1058-1059
- Robbins & Kumar Basic Pathology, p. [3259]
- Fitzpatrick's Dermatology (9th ed.), Vol. 1-2, pp. 1913-1914 (Chapter 111)
- Emery's Elements of Medical Genetics and Genomics
Recent evidence note (PubMed 2024): A 2024 review (PMID 39126021) proposes a dysembryogenetic theory of BCC pathogenesis - suggesting tumors arise from misplaced embryonic cell rests rather than mature basal cells - adding a complementary perspective to the established UV/Hedgehog model. This remains under investigation and does not replace current consensus.
Make it simple and I need it in 10 lines
Pathogenesis of Basal Cell Carcinoma (10 lines)
- BCC arises from the basal layer of the epidermis or outer root sheath of hair follicles.
- The most important trigger is chronic UV-B radiation (290-320 nm) causing DNA damage.
- UV-induced mutations cause C→T transitions - the UV signature - in key tumor suppressor genes.
- The primary target is PTCH1 (Patched-1), mutated in ~73% of sporadic BCCs.
- Normally, PTCH1 inhibits SMO (Smoothened); when PTCH1 is lost, SMO becomes constitutively active.
- Active SMO activates GLI1 transcription factors, driving unregulated cell proliferation.
- This constitutive activation of the Sonic Hedgehog (SHH) signaling pathway is the hallmark of ALL BCCs.
- TP53 mutations (found in ~50-61% of BCCs) impair apoptosis, allowing damaged cells to survive.
- Gorlin syndrome (inherited PTCH1 loss) confirms this pathway - patients develop hundreds of BCCs early in life.
- Additional risk factors - immunosuppression, ionizing radiation, and arsenic - amplify DNA damage and impair surveillance, compounding the risk.
Melanoma same 10 points of pathogenesis
Pathogenesis of Melanoma (10 lines)
-
Cell of origin: Melanoma arises from melanocytes - the pigment-producing cells in the basal layer of the epidermis and hair follicles.
-
UV radiation (especially intermittent intense sun exposure and severe sunburns early in life) is the most important environmental trigger, causing accumulating DNA mutations with the classic UV signature (C→T transitions) in melanocytes.
-
The most common driver mutation is BRAF V600E (~50% of cutaneous melanomas), a gain-of-function mutation that constitutively activates the MAPK/RAS-RAF-MEK-ERK proliferative signaling pathway.
-
CDKN2A (encoding p16 and ARF) is mutated in ~40% of familial melanomas - loss of p16 releases CDK4/6 to drive cell cycle progression, and loss of ARF destabilizes p53, disabling apoptosis.
-
The PI3K/AKT/mTOR pathway is constitutively activated (via PTEN loss or AKT mutations) in most melanomas, promoting cell survival and resistance to apoptosis.
-
TERT (telomerase) promoter mutations are among the most common mutations in sporadic melanoma, enabling unlimited replicative potential by maintaining telomere length.
-
NF1 loss-of-function mutations (~14%) remove a RAS-GAP brake, leading to constitutive RAS activation and proliferative signaling - defining a third genomic subtype.
-
Melanoma progresses in defined stages: radial growth phase (horizontal spread, low metastatic potential) → vertical growth phase (invasion into dermis, high metastatic potential); this transition is the critical step in lethality.
-
Immune evasion plays a major role - melanoma upregulates PD-L1, downregulates MHC class I, and recruits immunosuppressive cells, allowing escape from T-cell surveillance (the basis of checkpoint inhibitor therapy).
-
Germline predisposition (autosomal dominant CDKN2A mutations, CDK4 mutations, MC1R variants causing fair skin/red hair) accounts for ~10% of cases, with the remaining 90% being sporadic and UV-driven.
Make ckdn2a clear flow chart
A clean, clear medical flowchart titled "CDKN2A Mutations in Melanoma Pathogenesis" showing two parallel pathways side by side. LEFT PATHWAY titled "p16 (INK4a) Arm": - Box: "Normal CDKN2A gene → produces p16 protein" - Arrow down - Box: "p16 INHIBITS CDK4/CDK6" - Arrow down - Box: "CDK4/6 cannot phosphorylate RB protein" - Arrow down - Box: "RB remains ACTIVE → blocks E2F transcription factor" - Arrow down - Box: "Cell stays in G1 phase → NO proliferation" - Then a RED X branch showing: "CDKN2A mutation → p16 LOST → CDK4/6 unblocked → RB phosphorylated/inactivated → E2F released → Cell enters S phase → Uncontrolled proliferation → MELANOMA" RIGHT PATHWAY titled "ARF (p14ARF) Arm": - Box: "Normal CDKN2A gene → produces ARF protein" - Arrow down - Box: "ARF INHIBITS MDM2 (an E3 ubiquitin ligase)" - Arrow down - Box: "MDM2 cannot degrade p53" - Arrow down - Box: "p53 remains ACTIVE → triggers cell cycle arrest or apoptosis" - Arrow down - Box: "Damaged cells are eliminated" - Then a RED X branch showing: "CDKN2A mutation → ARF LOST → MDM2 active → p53 degraded → No apoptosis → Damaged melanocytes survive → MELANOMA" At the bottom, a large red convergence box: "CDKN2A LOSS = Double hit: loss of RB pathway AND loss of p53 pathway → Melanocyte immortalization and melanoma development" Use white background, blue boxes for normal pathway, red boxes for mutant/cancer pathway, clear black arrows, bold labels. Professional medical diagram style.
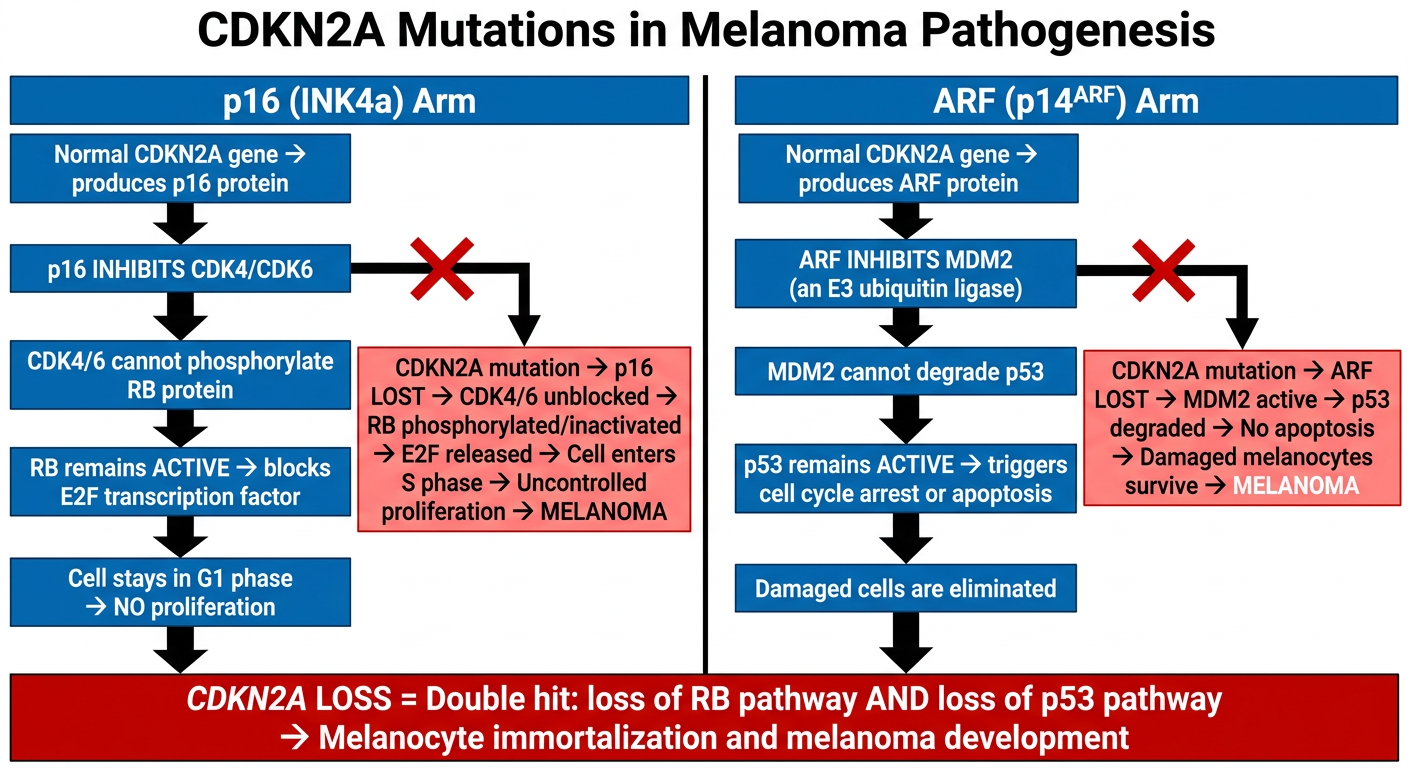
- When CDKN2A is lost: CDK4/6 runs free → RB inactivated → cell divides uncontrollably
- When CDKN2A is lost: MDM2 degrades p53 freely → damaged melanocytes escape death