Explain the mechanism and regulation of HCl secretion. Describe the protective role of “gastric mucosal barrier.” Add a note on the physiological basis of treatment of gastric ulcer.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Searching PubMed
peptic ulcer disease treatment guidelines proton pump inhibitor
I now have all the material I need to write a thorough, well-sourced answer. Let me compose the full response.
HCl Secretion: Mechanism, Regulation, Gastric Mucosal Barrier, and Treatment of Peptic Ulcer
1. Mechanism of HCl Secretion
Site and Cell of Origin
HCl is secreted exclusively by parietal cells (oxyntic cells) located in the oxyntic glands of the fundus and body of the stomach (the proximal ~80%). These glands also contain chief cells (pepsinogen), mucous neck cells, and ECL cells (histamine). The secreted acid solution contains approximately 160 mmol/L HCl, with a pH of ~0.8 - meaning the H+ concentration is about 3 million times that of arterial blood. Generating this gradient requires over 1500 calories of energy per litre of gastric juice.
Intracellular Canaliculi
Each parietal cell contains large branching intracellular canaliculi lined with villus-like projections where the actual acid is formed. From these canaliculi, HCl is conducted to the secretory end of the cell and discharged into the gland lumen.
Ionic Steps of HCl Formation
The following figure from Guyton illustrates the full mechanism:
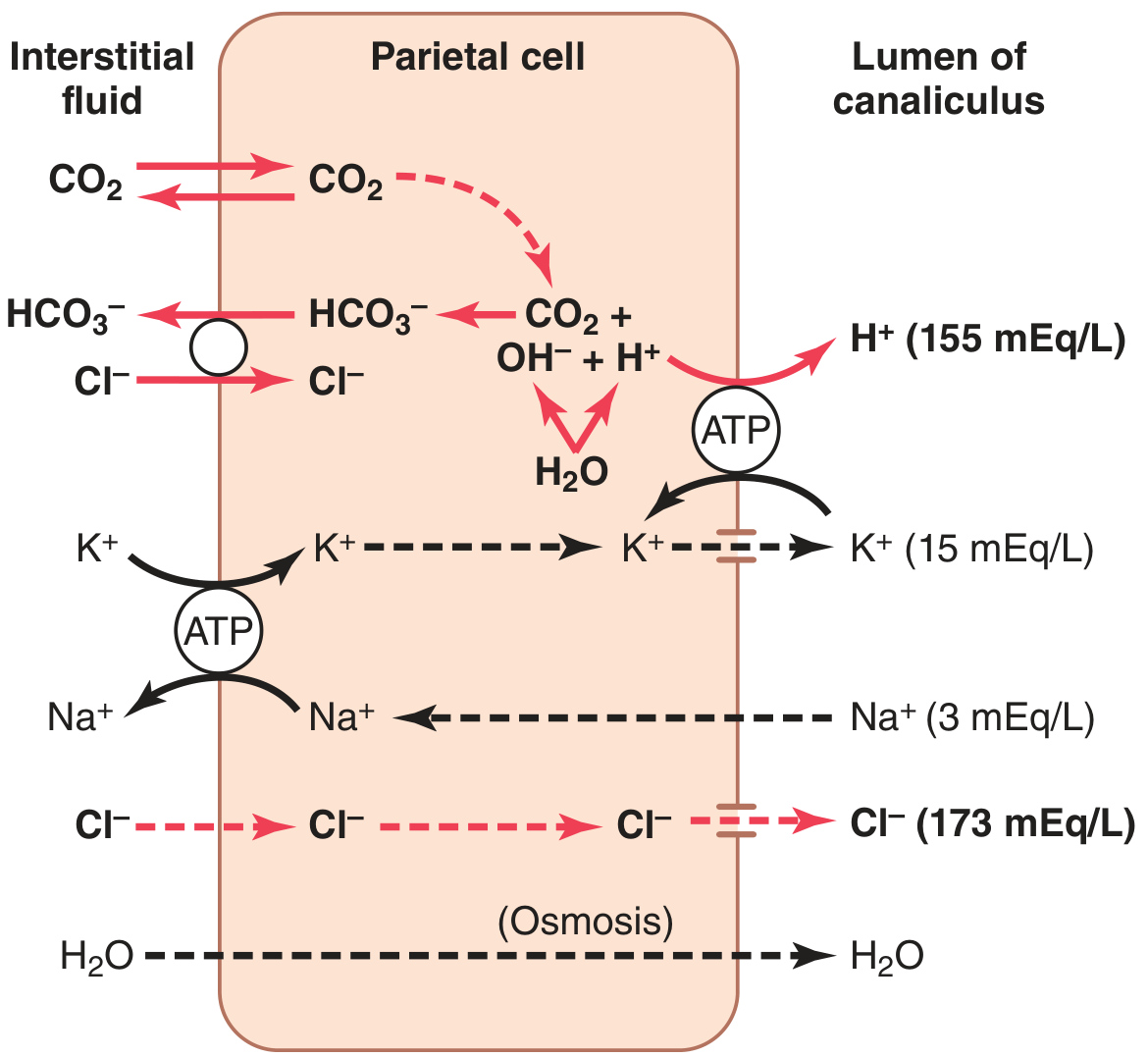
Figure 65.6 - Guyton & Hall Textbook of Medical Physiology
Step-by-step:
-
Water dissociation: Inside the parietal cell, H2O dissociates into H+ and OH-.
-
H+-K+ ATPase (proton pump): The H+ is actively pumped into the canalicular lumen in exchange for K+ via the H+-K+ ATPase located on the apical (canalicular) membrane. This is the primary driving force of acid secretion and is the direct target of proton pump inhibitors (PPIs).
-
CO2 hydration: The OH- that accumulates inside the cell combines with CO2 (from cellular metabolism or from blood) to form HCO3-, a reaction catalyzed by carbonic anhydrase.
-
HCO3-/Cl- exchanger: HCO3- is transported across the basolateral membrane into the extracellular fluid (and then blood) in exchange for Cl-, which enters the cell. This is the "alkaline tide" - gastric venous blood has a higher pH than arterial blood during active secretion.
-
Cl- secretion: Cl- exits the cell through chloride channels into the canaliculus, meeting the secreted H+ to form HCl.
-
K+ recycling: K+ that entered via the H+-K+ ATPase is recycled back into the canalicular lumen through K+ channels, keeping the pump running.
-
Water: Follows by osmosis, producing a final secretion of H2O + HCl (~150-160 mEq/L) + KCl (~15 mEq/L) + small amounts of NaCl.
On the basolateral side, a Na+-K+ ATPase maintains the intracellular ionic environment by pumping Na+ out and K+ in.
2. Regulation of HCl Secretion
Three Primary Stimulants
HCl secretion is controlled by three main secretagogues acting on parietal cells, each binding specific receptors:
| Stimulant | Receptor on Parietal Cell | Source | Second Messenger |
|---|---|---|---|
| Acetylcholine (ACh) | Muscarinic M3 | Vagus nerve / enteric plexus | IP3/DAG (Ca2+) |
| Gastrin | CCK-B (gastrin) receptor | G cells of antrum | Ca2+ |
| Histamine | H2 receptor | ECL cells (paracrine) | cAMP |
All three pathways ultimately activate the H+-K+ ATPase, but they act synergistically - blocking one pathway reduces the response to the others.
The ECL Cell as the Central Relay
Parietal cells work in close association with enterochromaffin-like (ECL) cells in the oxyntic glands. Gastrin (from G cells) and ACh both stimulate ECL cells to release histamine, which then acts in a paracrine fashion directly on adjacent parietal cell H2 receptors. ECL-derived histamine is the most potent stimulant of acid secretion in humans.
Somatostatin: The Key Inhibitor
Somatostatin from antral D cells is the primary physiological brake on acid secretion. It inhibits acid secretion at three levels:
- Directly on parietal cells (D cell - parietal cell paracrine contact)
- On ECL cells (reduces histamine release)
- On G cells (reduces gastrin release)
Low antral pH stimulates somatostatin release - a negative feedback loop.
Phases of Gastric Secretion
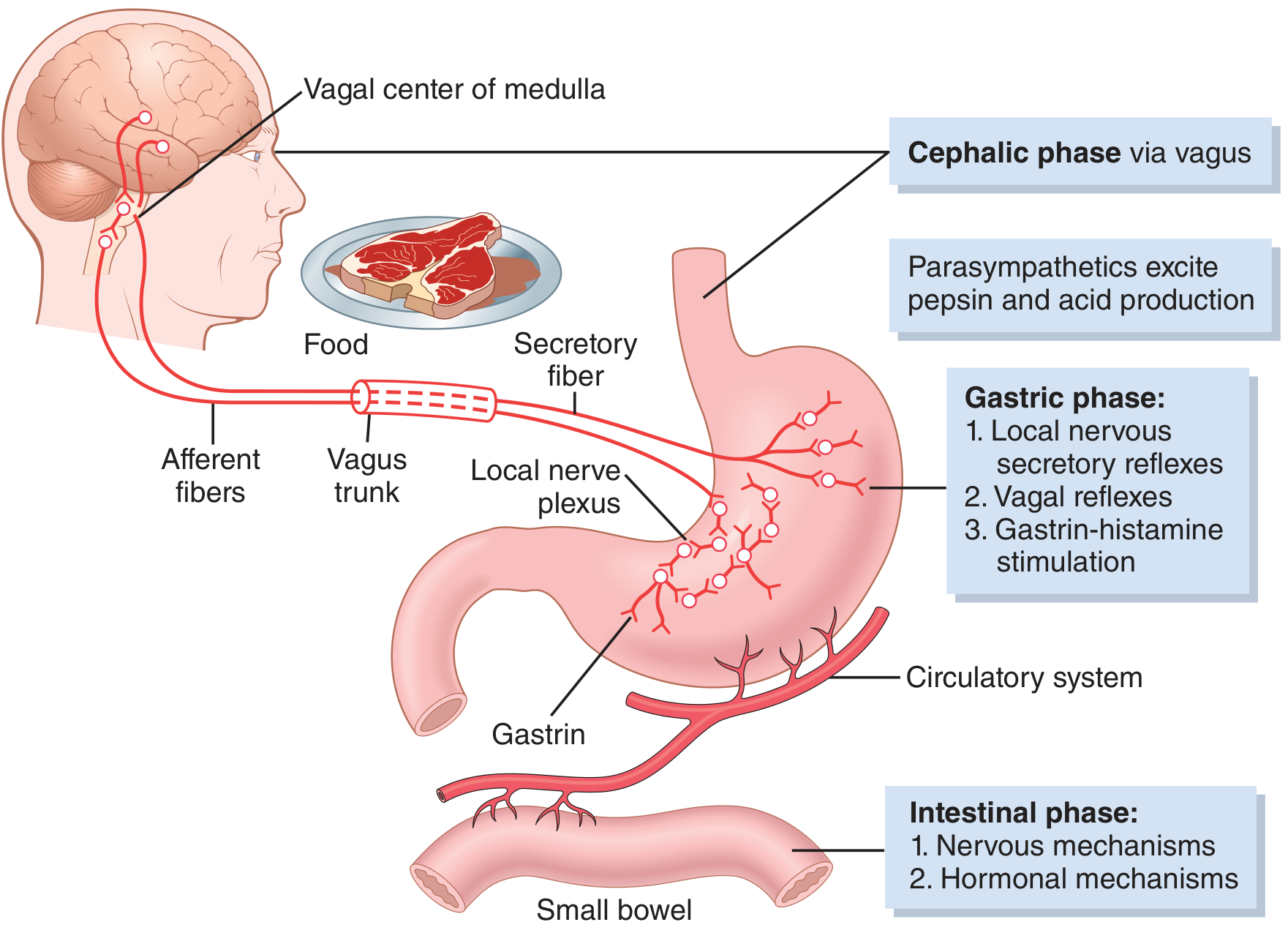
Figure 65.7 - Guyton & Hall Textbook of Medical Physiology
Cephalic Phase (~30% of meal response)
- Triggered by sight, smell, taste, or even thought of food
- Mediated via the cerebral cortex → hypothalamus/amygdala → dorsal motor nucleus of vagus → gastric vagal efferents
- ACh released directly stimulates parietal cells, chief cells, mucous cells, and G cells
Gastric Phase (~60% of meal response)
- Triggered by distension and protein content in the stomach
- Three mechanisms operate: (1) long vagovagal reflexes, (2) local enteric reflexes, (3) gastrin release from antral G cells
- This is the dominant phase and accounts for most of the ~1500 mL daily acid output
Intestinal Phase (~10%)
- Small amount of gastrin released by duodenal mucosa when chyme enters the duodenum
- Soon followed by inhibitory signals as the small intestine fills
Inhibitory Mechanisms (Intestinal Feedback)
Once chyme enters the small intestine, several mechanisms inhibit further secretion:
- Enterogastric reflex: Distension, acid, fat, or hyperosmolarity in the duodenum triggers a reverse reflex (via myenteric plexus and sympathetics) that inhibits gastric secretion
- Secretin: Released by S cells in response to duodenal acid; inhibits gastrin secretion and stimulates bicarbonate secretion
- GIP (glucose-dependent insulinotropic peptide), VIP, and somatostatin all have moderate inhibitory effects
3. The Gastric Mucosal Barrier
The stomach must protect itself from its own secretions - 160 mEq/L HCl and active pepsin. This protection is provided by a multi-layered gastric mucosal barrier.
Components (from Schwartz's Principles of Surgery)
| Layer | Mechanism |
|---|---|
| Mucus-bicarbonate layer | Viscid gel of mucopolysaccharides secreted by surface epithelial cells (SECs) and pyloric glands; contains HCO3- creating a favorable pH gradient (pH 7 near mucosa vs pH 2 in lumen) |
| Epithelial barrier | Intact cell membranes and tight junctions between epithelial cells prevent H+ from entering the interstitium |
| Hydrophobic phospholipids | Surfactant-like phospholipids on the mucosal surface repel aqueous acid |
| Restitution | Sloughed or denuded SECs are rapidly replaced by migration of adjacent cells |
| Mucosal blood flow (reactive hyperemia) | Rich submucosal blood supply removes any back-diffused H+; buffers acid with the alkaline tide from parietal cell HCO3- secretion |
| Afferent sensory neurons | Protective reflexes triggered by mucosal irritation; blocked by topical anesthetics |
Key Mediators of Cytoprotection
- Prostaglandins (PGE2, PGI2): Stimulate mucus and bicarbonate secretion, maintain mucosal blood flow, inhibit acid secretion. NSAIDs block prostaglandin synthesis via COX inhibition, which is why they are a major cause of mucosal injury.
- Nitric oxide (NO): Vasodilatory; maintains mucosal blood flow
- Epidermal growth factor (EGF): Promotes epithelial repair and restitution
- Calcitonin gene-related peptide (CGRP) and gastrin-releasing peptide (GRP): Protective neuropeptides
Barrier Breakers
The mucosal barrier is damaged by:
- NSAIDs / aspirin: Inhibit prostaglandin synthesis; allow back-diffusion of H+
- Alcohol: Direct mucosal damage and increased permeability
- Bile reflux: Disrupts the lipid layer
- H. pylori: Produces urease (generates NH3, which is locally toxic) and disrupts tight junctions and mucus production
- Ischaemia: The stomach is the most sensitive part of the GI tract to ischaemia and the slowest to recover after hypovolaemic shock - this explains the high incidence of stress ulceration
When barrier function fails, H+ back-diffuses into the lamina propria and submucosa. This triggers a protective increase in mucosal blood flow; if this response is also blocked (e.g., by shock or NSAIDs), gross mucosal ulceration results.
4. Physiological Basis of Treatment of Gastric Ulcer
Gastric ulceration reflects an imbalance between aggressive factors (HCl, pepsin, H. pylori, NSAIDs, bile) and defensive factors (mucus, bicarbonate, blood flow, prostaglandins). Treatment targets this imbalance.
A. Reducing Acid Secretion
1. Proton Pump Inhibitors (PPIs) - e.g., omeprazole, esomeprazole, pantoprazole
- Target the final common pathway - the H+-K+ ATPase on the parietal cell apical membrane
- PPIs are prodrugs that become activated in the acidic canalicular environment, forming a covalent bond with cysteine residues on the pump - providing long-lasting inhibition (irreversible until new pump protein is synthesized)
- Most effective acid suppression available; the cornerstone of ulcer treatment
- Omeprazole 20 mg twice daily is equivalent to standard PPI dosing for complicated ulcer disease
2. H2-Receptor Antagonists (H2RAs) - e.g., ranitidine, famotidine
- Block histamine's action at H2 receptors on parietal cells, reducing cAMP-mediated acid secretion
- Less powerful than PPIs (histamine is not the only stimulant) but useful for maintenance or milder disease
3. Anticholinergic agents / Vagotomy
- Block muscarinic M3 receptors or cut the vagal drive to parietal cells and G cells
- Surgical vagotomy (truncal or highly selective) reduces both cephalic and gastric phase acid output
- Highly selective vagotomy spares the hepatic and celiac branches, preserving gastric emptying
4. Antacids (e.g., aluminium hydroxide, magnesium hydroxide)
- Directly neutralize secreted HCl in the lumen: HCl + NaHCO3 → NaCl + H2O + CO2
- Raise intragastric pH, inactivating pepsin (inactive above pH 5)
- Provide rapid symptomatic relief but no mucosal healing; used adjunctively
B. Eradicating H. pylori
- H. pylori infects the gastric mucosa, induces chronic inflammation, disrupts the mucosal barrier, and increases gastrin secretion
- Triple therapy: PPI + clarithromycin + amoxicillin (or metronidazole) for 10-14 days eradicates the organism
- Eradication dramatically reduces ulcer relapse rates (from ~70-80% to <10% at 1 year)
C. Enhancing Mucosal Defense
1. Sucralfate
- A sucrose-sulfate-aluminium complex that binds to ulcer craters, forming a protective coating
- Stimulates prostaglandin synthesis and EGF release; buffers acid locally
- Acts independently of acid suppression - a pure "mucosal defense enhancer"
2. Bismuth salts
- Coat ulcer base; also have direct anti-H. pylori activity
- Part of quadruple therapy regimens
3. Prostaglandin analogues (e.g., misoprostol)
- Exogenous PGE1 analogue: stimulates mucus and bicarbonate secretion, reduces acid secretion, maintains mucosal blood flow
- Used particularly to prevent NSAID-induced ulcers (where endogenous prostaglandins are depleted)
D. Avoiding Ulcerogenic Factors
- Withdraw NSAIDs if possible; use COX-2-selective inhibitors (lower GI risk) with PPI cover if NSAIDs are essential
- Cease alcohol and smoking (smoking reduces bicarbonate secretion and delays ulcer healing)
Summary Table
| Mechanism | Drug/Intervention |
|---|---|
| Irreversible H+-K+ ATPase block | PPIs (omeprazole, pantoprazole) |
| H2 receptor block | Ranitidine, famotidine |
| Acid neutralization | Antacids (MgOH, AlOH) |
| Muscarinic block / vagotomy | Pirenzepine; surgical vagotomy |
| H. pylori eradication | Triple/quadruple therapy |
| Mucosal coating / cytoprotection | Sucralfate, bismuth |
| Prostaglandin replacement | Misoprostol |
| Reduce NSAID-induced injury | COX-2 inhibitors + PPI |
Sources:
- Guyton and Hall Textbook of Medical Physiology (14th ed.), pp. 802-804
- Schwartz's Principles of Surgery (11th ed.) - Gastric Mucosal Barrier (Table 26-3)
- Bailey and Love's Short Practice of Surgery (28th ed.) - Gastric mucus and the gastric mucosal barrier
- Sabiston Textbook of Surgery - Stimulated Acid Secretion; Other Gastric Secretory Products
- Goldman-Cecil Medicine - Medical Therapy for Peptic Ulcer Disease
This is a shared conversation. Sign in to Orris to start your own chat.